Инсулин принимает участие в регуляции метаболизма, трансмембранного переноса ионов, аминокислот. Влияние инсулина на углеводный обмен трудно переоценить. У людей с сахарным диабетом также проявляются признаки нарушений различных видов метаболизма.
Сахарный диабет в последнее время диагностируется все больше. Заболевания вызывает различные нарушения обмена веществ. Сахарный диабет, патологическая физиология которого может сильно различаться, находится на третьем месте после онкологии и сердечно-сосудистых заболеваний. В мире насчитывается около 100 миллионов людей, больных диабетом. Каждые 10 лет количество диабетиков становится больше в 2 раза.
Самому большому риску заболеть подвержены люди из развивающихся стран и маргинальные элементы в развитых государствах. Нарушение обмена веществ при сахарном диабете, ведет к различным патологиям. Диабетом 2 типа чаще заболевают люди после 45 лет.
 В 1869 году Лангерганс нашел в поджелудочной железе островки, которые позже назвали его именем. Стало известно, что сахарный диабет может появиться после удаления железы.
В 1869 году Лангерганс нашел в поджелудочной железе островки, которые позже назвали его именем. Стало известно, что сахарный диабет может появиться после удаления железы.
Инсулин это белок, то есть полипептид, который состоит из А и Б цепей. Они соединены двумя дисульфидными мостами. Сейчас известно, что инсулин формируется и запасается бета-клетками. Инсулин нарушается под воздействием ферментов, которые восстанавливают дисульфидные связи и носят название «инсулиназа». Далее протеолитические ферменты занимаются гидролизацией цепей до низкомолекулярных частей.
Считается, что основным ингибитором секреции инсулина выступает сам инсулин в крови, и еще гипергликемические гормоны:
ТТГ, катехоламины, АКТГ, СТГ и глюкагон разными путями активируют в клеточной мембране аденилциклазу. Последняя активизирует формирование циклического 3,5 аденозин-монофосфата, он активирует другое элемент – протеинкиназу, она фосфолирирует микротрубочки бета-островков, что ведет к замедлению высвобождения инсулина.
Микротрубочки являются каркасом бета-клетки, по которой раньше синтезированный инсулин двигается в везикулах к клеточной мембране.
Самым сильным стимулятором формирования инсулина является глюкоза крови.
Механизм действия инсулина заключается также в антагонистических отношениях внутриклеточных посредников 3,5 – ГМФ и 3,5 АМФ.
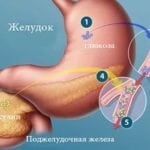 Инсулин воздействует на обмен углеводов при сахарном диабете. Ключевым звеном при этом заболевании является недостаточность данного вещества. Оказывается большое влияние инсулина на обмен углеводов, а также на другие виды обменов, поскольку уменьшается секреция инсулина, снижается его активность либо нарушается рецепция клетками инсулинзависимых тканей.
Инсулин воздействует на обмен углеводов при сахарном диабете. Ключевым звеном при этом заболевании является недостаточность данного вещества. Оказывается большое влияние инсулина на обмен углеводов, а также на другие виды обменов, поскольку уменьшается секреция инсулина, снижается его активность либо нарушается рецепция клетками инсулинзависимых тканей.
Вследствие нарушения углеводного обмена при сахарном диабете понижается активность поступления глюкозы в клетки, возрастает ее объем в крови и активируются способы усвоения глюкозы, которые не зависят от инсулина.
Сорбитоловый шунт это состояние, когда глюкоза восстанавливается в сорбит, после чего окисляется во фруктозу. Но окисление ограничено инсулинзависисым ферментом. При активизации полиолового шунта возникает накопление в тканях сорбитола, это способствует появлению:
Происходит внутреннее образование глюкозы из белка и гликогена, но и этот вид гоюкозы клетки не усваивают, поскольку есть недостаток инсулина. Аэробный гликолиз и пентозофосфатный шунт угнетаются, появляется гипоксия клеток и энергетическая недостаточность. Возрастает объем гликированного гемоглобина, он не является носителем кислорода, что усиливает гипоксию.
Белковый обмен при сахарном диабете может быть нарушен:
- гиперазотемией (увеличение уровня остаточного азота),
- гиперазотемией (увеличение объема азотистых соединений в крови).
Норма азота белка – 0,86 ммоль/л, а общий азот должен составлять 0,87 ммоль/л.
Причинами патофизиологии становятся:
- увеличение катаболизма белка,
- активизиация дезаминирования аминокислоты в печени,
- остаточный азот.
Это происходит вследствие усиления деструкции белков, в основном, в печени и мышцах.
В моче при сахарном диабете увеличивается объем азотистых соединений. Азотурия имеет следующие причины:
- увеличение концентрации продуктов с азотом в крови, их секреция с мочой,
- нарушенный жировой обмен характеризуется кетонемией, гиперлипидемией, кетонурией.
При диабете развивается гиперлипидемия, что является увеличением объема в крови уровня липидов. Их количество больше нормы, то есть больше 8 г/л. Существуют следующие гиперлипидемии:
- активация в тканях липолиза,
- торможение уничтожения липидов клетками,
- усиление синтеза холестерина,
- торможение доставки ВЖК в клетки,
- понижение активности ЛПЛазы,
- кетонемия – увеличение объема в крови кетоновых тел.
- ацетон,
- ацетоуксусная кислота,
- р-оксималярная кислота.
Суммарный объем кетоновых тел в крови может быть выше 30-50 мг%. На это есть такие причины:
- активация липолиза,
- усиление окисления в клетках ВЖК,
- приостановка синтеза липидов,
- снижение окисления ацетил — КоА в гепатоцитах с формированием кетоновых тел,
Выделение кетоновых тел вместе с мочой это проявления сахарного диабета неблагоприятного течения.
- много кетоновых тел, которые проходят фильтрацию в почках,
- нарушения водного обмена при сахарном диабете, проявляющееся полидипсией и полиурией,
Полиурия это патология, которая выражена в образовании и выделении мочи в объеме, который превышает нормальные показатели. В обычных условиях выделяется от 1000 до 1200 мл за одни сутки.
При сахарном диабете суточный диурез составляет 4000-10 000 мл. Причинами является:
- Гиперосмия мочи, которая возникает из-за выведения избытка глюкозы, ионов, КТ и азотистых соединений. Таким образом, стимулируется фильтрация жидкости в клубочках и тормозит реабсорцию,
- Нарушение реабсорбции и экскреции, которые вызваны диабетической невропатией,
- Полидипсия.
 Печень под воздействием инсулина может запасать только определенный объем гликогена. Излишки глюкозы, которая поступила в печень, начинает фосфорилироваться и таким образом удерживаться в клетке, но затем они трансформируются в жир, а не в гликоген.
Печень под воздействием инсулина может запасать только определенный объем гликогена. Излишки глюкозы, которая поступила в печень, начинает фосфорилироваться и таким образом удерживаться в клетке, но затем они трансформируются в жир, а не в гликоген.
Эта трансформация в жир выступает результатом прямого воздействия инсулина, а формирующиеся при этом жирные кислоты кровь транспортирует в жировую ткань. В крови жиры входят в состав липопротеинов, которые играют важную роль в формировании атеросклероза. Вследствие этой патологии может начаться:
Действие инсулина на клетки жировой ткани похоже на его действие на клетки печени, но в печени формирование жирных кислот проходит более активно, поэтом они переносятся из нее в жировую ткань. Жирные кислоты в клетках хранятся в виде триглицеридов.
Под воздействием инсулина снижается распад триглицеридов в жировой ткани, благодаря ингибированию липазы. Кроме этого, инсулин активизирует синтез жирных кислот клетками и участвует в их снабжении глицеролом, который нужен для синтеза триглицеридов. Таким образом, со временем накапливается жир, в том числе и в этом заключается физиология сахарного диабета.
Действие инсулина на жировой обмен может быть обратимо, при его низком уровне триглицериды вновь расщепляются на жирные кислоты и глицерол. Это связано с тем, что инсулин ингибирует липазу и при снижении его объема активируется липолиз.
Жирные свободные кислоты, которые формируются при гидролизе триглицеридов, одновременно поступают в кровь и применяются как источник энергии для тканей. Окисление этих кислот может быть во всех клетках, исключая нервные.
Большее количество жирных кислот, которые освобождаются при нехватке инсулина из жировых блоков, вновь поглощается печенью. Клетки печени могут синтезировать триглицериды и при отсутствии инсулина. При нехватке этого вещества, освобождающиеся из блоков жирные кислоты, собираются в печени в триглицеридовом виде.
По этой причине у людей с нехваткой инсулина, несмотря на общую тенденцию к сбросу веса, формируется ожирение печени.
 При диабете инсулинглюкагоновый индекс понижен. Это объясняется снижением секреции инсулина, а также увеличением выработки глюкагона.
При диабете инсулинглюкагоновый индекс понижен. Это объясняется снижением секреции инсулина, а также увеличением выработки глюкагона.
Нарушения липидного обмена при сахарном диабете выражается в слабой стимуляции складирования и увеличении стимуляции мобилизации запасов. После употребления еды, в режиме постабсорбтивного состояния находятся:
Продукты переваривания и их метаболиты, вместо того, чтобы откладываться как жиры и гликоген, циркулируют в крови. В определенной мере возникают и циклические процессы, например, одновременно протекающие процессы глюконеогенеза и гликолиза, а также процесс распада жиров и синтеза.
Все формы сахарного диабета отличаются сниженной толерантностью к глюкозе, то есть, гиперглюкоземия после употребления пищи или даже на голодный желудок.
Основными причинами гиперглюкоземии является:
- употребление жировой тканью и мышцами ограничено, поскольку при отсутствии инсулина ГЛБТ-4 не экспонируется на поверхности адипоцитов и миоцитов. Глюкоза не может быть запасена в виде гликогена,
- глюкоза в печени не применяется для запасания в виде гликогена, поскольку при низком объеме инсулина и высокого объема глюкагона гликогенсинтаза пребывает в неактивной форме,
- глюкоза в печени не используется для синтеза жиров. Ферменты гликолиза и пируватдегидрогеназа пребывают в пассивной форме. Заторможено превращение глюкозы в ацетил -СоА, который нужен для синтеза жирных кислот,
- путь глюконеогенеза при небольшой концентрации инсулина и высокой глюкагона активирован и возможен синтез глюкозы из глицерина и аминокислот.
Еще одним характерным проявлением сахарного диабета выступает повышенный уровень в крови липопротеинов, кетоновых тел и свободных жирных кислот. Пищевые жиры не депонируются в жировой ткани, поскольку липаза адипоцитов пребывает в активной форме.
Появляется высокое содержание жирных свободных кислот в крови. Жирные кислоты поглощает печень, их часть трансформируется в триацилглицерины, они в составе ЛОНП попадают в кровь. Определенное количество жирных кислот вступает в β-окисление в митохондриях печени, а формирующийся ацетил-СоА применяется для синтеза кетоновых тел.
Влияние инсулина на обмен веществ заключается также в том, что при введении инсулина в разных тканях организма происходит ускорение синтеза жиров и распад триглицеридлипидов. Нарушение обмена липидов это запасание жира, который служит удовлетворению энергетических потребностей при неблагоприятных ситуациях.
Чрезмерное появление цАМФ ведет к снижению синтеза белков и понижению ЛВП и ЛОНП. В результате снижения ЛВП понижается выведение холестерина из мембран клеток в плазму крови. Холестерин начинает откладываться в стенках мелких сосудов, что приводит к формированию диабетической ангиопатии и атеросклероза.
В результате снижения ЛОНП – в печени накапливается жир, он в норме выводится в составе ЛОНП. Белковый синтез подавляется, что вызывает понижение формирования антител, а затем, и недостаточную устойчивость больных диабетом к инфекционным заболеваниям. Известно, что люди, имеющие нарушения белкового обмена страдают фурункулезом.
 Микроангиопатия представляет собой диабетический гломерулонефрит. Из-за диабетической ретинопатии, люди с сахарным диабетом теряют зрение в 70-90% случаев. В частности, у диабетиков развивается катаракта.
Микроангиопатия представляет собой диабетический гломерулонефрит. Из-за диабетической ретинопатии, люди с сахарным диабетом теряют зрение в 70-90% случаев. В частности, у диабетиков развивается катаракта.
Вследствие нехватки ЛВП возникает избыточный холестерин в клеточных мембранах. Поэтому может появиться ишемическая болезнь сердца или облитерирующий эндартериит. Вместе с этим формируется микроангиопатия с нефритом.
При диабете формируется пародонтоз с гингивитом – пародонтитом – пародонтозом. У диабетиков нарушаются структуры зубов и поражаются опорные ткани.
Причинами патологии микрососудов в этих случаях, скорее всего, является формирование необратимых сшивок глюкозы с белками стенки сосудов. Тромбоциты при этом выделяют фактор, который стимулирует рост гладкомышечных составляющих сосудистой стенки.
Нарушения жирового обмена выражаются также в том, что жировая инфильтрация печени увеличивается в печени ресинтез липидов. В норме их выводят в виде ЛОНП, формирование которых зависит от объема белка. Для этого нужны донаторы группы СНЗ, то есть холин или метионин.
Синтез холина стимулирует липокаин, который вырабатывается эпителием протоков поджелудочной. Нехватка его приводит к ожирению печени и формированию тотального и островкового видов диабета.
Инсулиновая недостаточность ведет к низкой устойчивости к инфекционным заболеваниям. Таким образом, формируется фурункулез.
О влиянии инсулина на организм расскажет видео в этой статье.
У людей с сахарным диабетом (СД) значительно ухудшаются обменные процессы в организме, что повышает риск смертности в несколько раз. Значимым, но не единственным спутником СД, является нарушенный углеводный обмен. На фоне дефицита инсулина концентрация липидов в сыворотке крови критически повышается. У людей с СД 2 типа часто наблюдаются болезни сердечно-сосудистой системы, причем при повышенных липидах даже в условии компенсации диабета купировать процесс макрососудистых нарушений сложно. Улучшить состояние позволит только комплексный подход в лечении на фоне постоянного контроля сахара, АД (давления) и количества липидов в крови.
ВАЖНО ЗНАТЬ! Даже «запущенный» диабет можно вылечить дома, без операций и больниц. Просто прочитайте что говорит Марина Владимировна читать рекомендацию.

Липиды — естественные жиры, которые синтезируются в клетках печени. Основным их источником считаются продукты питания. Большинство из них не растворяются в крови, поэтому транспортируются к клеткам органов, прикрепляясь к белкам. Липидный обмен считается одним из самых сложных физиологических процессов, которые происходят в организме человека. В его функции входит обмен холестерина, фосфолипидов и триацилглицеролов, которые транспортируются с кишечника в органы.
Сахар снижается мгновенно! Диабет со временем может привести к целому букету заболеваний, таких как проблемы со зрением, состоянием кожи и волос, появлению язв, гангрены и даже раковых опухолей! Люди, наученные горьким опытом для нормализации уровня сахара пользуются. читать далее.
Нарушение обмена липидов при сахарном диабете распространенное явление. В норме клетки печени беспрепятственно принимают глюкозу. Под действием разрушительных процессов нарушается выработка инсулина, что приводит к торможению выработки гексокиназы (фермента, участвующего в окислении глюкозы). А это чревато нарушением усваивания сахарных молекул. Неправильный биосинтез ферментов, вызванный инсулиновой недостаточностью, провоцирует нарушение белкового обмена, сбоем в процессах расщепления жиров.
Биохимия липидной дисфункции проявляется стремительным повышением содержания глюкозы в крови.
На фоне недостаточного усвоения сахара при дефиците инсулина наблюдаются ускоренный распад жиров, повышение уровня кетоновых тел при замедлении выработки жирных кислот и триацилглицеролов. Нарушение синтеза липидов при сахарном диабете провоцирует следующие патологические состояния:
- ожирение;
- увеличение объемов печени и селезенки;
- появление очаговых кожных образований в области глаз и сухожилия;
- повышение уровня холестерина в крови;
- нарушение работы почек;
- повышение артериального давления.
Вернуться к оглавлению
Различают 2 вида нарушения липидного обмена: первичное и вторичное. Первый развивается вследствие генетических мутаций, при которых отмечается гиперактивная выработка холестерина или снижение активности его утилизации. Вторичное нарушение провоцируется неправильным образом жизни, злоупотреблением калорийной и жирной пищи, а также множеством сопутствующих заболеваний.
Липидный обмен при сахарном диабете нужно постоянно контролировать. Для этого используют специальные лабораторные исследования, которые помогут увидеть полную картину нарушения синтеза и усваивания жиров. В первую очередь нужно провести липидограмму, которая определяет количественное соотношение всех липидных классов. Для более точной диагностики проводят общий анализ крови и липопротеидограмму. Чтобы исключить развитие патологии сердечно-сосудистой системы рекомендуется обратиться за консультацией к кардиологу и при необходимости пройти дополнительное обследование.
Нарушение липидного обмена при сахарном диабете подлежит немедленной коррекции. В первую очередь используют немедикаментозные способы лечения:
- соблюдение диеты с низким содержанием животных жиров и углеводов;
- повышение двигательной активности;
- исключение алкогольных напитков и кофе;
- отказ от вредных привычек;
- нормализация психоэмоционального состояния.
Медикаментозное лечение липидного нарушения проводится только в том случае, если диетотерапия и коррекция образа жизни не принесли должного результата. В обязательном порядке назначаются препараты, которые способствуют угнетению синтеза холестерина в организме. А также основную терапию дополняют биологически активными добавками с высоким содержанием таурина, который не только нормализует и поддерживает липидный обмен, но и благоприятно влияет на динамику лечения сахарного диабета.
Судя по тому, что вы сейчас читаете эти строки — победа в борьбе с высоким уровнем сахара в крови пока не на вашей стороне.
И вы уже думали о стационарном лечении? Оно и понятно, ведь диабет — очень опасное заболевание, которое при несвоевременном лечении может закончиться летальным исходом. Постоянная жажда, учащенное мочеиспускание, нечёткость зрения. Все эти симптомы знакомы вам не понаслышке.
Но возможно правильнее лечить не следствие, а причину? Рекомендуем прочитать статью о современных методах лечения диабета. Читать статью >>
НАРУШЕНИЯ УГЛЕВОДНОГО ОБМЕНА в основном заключаются в следующем:
1) затруднение транспорта глюкозы в мышечную и жировую ткань;
2) угнетение окисления глюкозы по пути фосфорилирования в связи со снижением активности ключевых ферментов, превращения глюкозы (гексокиназы, гликокиназы);
3) понижение синтеза гликогена в печени в связи со снижением активности гликокиназы;
Следствием всех этих прцессов является развитие основного симптома сахарного диабета – гипергликемии.
Основной путь превращения глюкозы в физиологических условиях – это путь окислительного фосфорилирования, осуществляемый под действием инсулина. В условиях его дефицита процесс окисления глюкозы по пути фосфорилирования угнетается и увеличивается удельный вес других путей обмена глюкозы. В частности, начинает преобладать анаэробное расщепление глюкозы. В результате в тканях образуется в повышенном количестве молочная кислота. Выделение ее в кровь ведет к развитию гиперлактацедемии, усугубляющей диабетический ацидоз. Кроме того, усиливается превращение глюкозы по сорбитоловому пути и, соответственно, накапливаются продукты этого превращения (сорбитол и фруктоза).
Сорбитоловый путь превращения глюкозы характерен для хрусталика глаза, нервной ткани, эндотелия сосудов. Накопление сорбитола и фруктозы в тканях способствует развитию осложнений сахарного диабета (катаракта, полинейропатия, ангиопатия).
При диабете увеличивается также использование глюкозы в образовании гликопротеидов (белков, составляющих базальную мембрану сосудов), что играет важную роль в патогенезе микроангиопатий.
Гипергликемия вызывает гликозилирование различных белков: гемоглобин, альбумин, белки базальной мембраны сосудов, что приводит к изменению их свойств, повышению иммуногенности и имеет значение в развитии сосудистых поражений.
Повышение уровня глюкозы в крови выше почечного порога (9,5-10 ммоль/л) сопровождается выделением сахара с мочой – глюкозурией, которая тем выше, чем интенсивнее гипергликемия. Выделение глюкозы с мочой сопроваждается увеличением диуреза. Глюкоза увлекает за собой жидкость в связи с повышением осмотического давления в провизорной моче и снижением реабсорбции мочи в канальцах почек. На каждый грамм глюкозы выделяется 20-40 мл жидкости. Таков механизм полиурии. Полидипсия при диабете вторичного происхождения. Она связана с интенсивным обезвоживанием организма.
Одновременно нарушается ВОДНО-СОЛЕВОЙ ОБМЕН. Больной теряет калий и натрий, усиление диуреза приводит к дегидратации.
НАРУШЕНИЕ ЖИРОВОГО ОБМЕНА при дефиците инсулина сводится к снижению синтеза жира и усилению липолиза. В результате мобилизации жира из депо развивается гиперлипидемия. Избыточный жир откладывается в бедных гликогеном гепатоцитах, вызывая жировую инфильтрацию печени. В кровь выделяются в повышенных количествах неэстерифицированные жирные кислоты, заменяющие глюкозу в качестве энергетического материала.
В печени, в условиях пониженного содержания гликогена, уменьшено превращение ацетил-Кол, а в цикле Кребса образуются в повышенном количестве недоокисленные продукты жирового обмена – кетоновые тела (оксимасляная кислота, ацетоуксусная кислота, ацетон). Развивается характерный для декомпенсации сахарного диабета кетоацидоз.
НАРУШЕНИЕ БЕЛКОВОГО ОБМЕНА заключается в угнетении синтеза белка и повышенном его распаде. Вследствие угнетения синтеза белка в детском возрасте наблюдается задержка роста.
В печени белок интенсивно превращается в углеводы. В процессе этого превращения диспротеинемии в сторону преобладания глобулиновых фракций образуются продукты распада (аммиак, мочевина, аминокислоты). Поступая в кровь, они обуславливают гиперазотемию.
При сахарном диабете вследствие нарушения белкового обмена снижается продукция защитных белков, что приводит к снижению иммунитета. Компенсация нарушений углеводного обмена, как правило, значительно улучшает показатели белкового, жирового и водно-солевого обмена. Однако нередко в клинической практике приходится прибегать к специальному лечению этих нарушений.
Не нашли то, что искали? Воспользуйтесь поиском:
Лучшие изречения: Студент — человек, постоянно откладывающий неизбежность. 10140 —  | 7196 —
| 7196 —  или читать все.
или читать все.
178.45.150.72 © studopedia.ru Не является автором материалов, которые размещены. Но предоставляет возможность бесплатного использования. Есть нарушение авторского права? Напишите нам | Обратная связь.
Отключите adBlock!
и обновите страницу (F5)
очень нужно
Основные патогенетические факторы метаболического повреждения включают следующие процессы:
1. Неферментативное гликозилирование белков — соединение глюкозы со свободными аминогруппами белков — коллагенов, кристаллинов, гемоглобина и др.
2. Ферментативное гликозилирование заключается в повышении скорости превращения глюкозы в сорбитол и маннитол, что приводит к их накоплению в клетках и межклеточном веществе сетчатки, хрусталика, клубочках почек, эндотелиоцитах, шванновских клетках, нейронах.
3. Резкие суточные колебания осмотического давления крови (из-за резкого повышения концентрации глюкозы после приема пищи).
4. Формирование внутриклеточной гиперосмолярности (за счет образования сорбитола).
5. Повреждение свободными радикалами («окислительный стресс»).
Патогенез диабетического кетоацидоза
Ведущие патогенетические звенья диабетического кетоацидоза, учитывающие нарушения всех видов обмена, представлены на интегральной схеме (рис. 12-28).
Среди важнейших метаболических нарушений при СД выделяются следующие:
1. Нарушения углеводного обмена при отсутствии инсулина (или его действия) характеризуются гипергликемией, глюкозурией и гиперлактатацидемией.
 Рис. 12-28. Патогенез диабетического кетоацидоза. Верхний ряд показывает основные метаболические эффекты при недостатке инсулина, которые вызывают все последующие изменения
Рис. 12-28. Патогенез диабетического кетоацидоза. Верхний ряд показывает основные метаболические эффекты при недостатке инсулина, которые вызывают все последующие изменения
• нарушением поступления глюкозы из крови внутрь клеток;
• компенсаторным ускорением гликогенолиза;
• активацией глюконеогенеза вследствие снятия репрессивного действия инсулина на синтез ключевых ферментов этого метаболического пути;
• усилением секреции глюкокортикоидов, являющихся стимуляторами синтеза ключевых ферментов глюконеогенеза в печени и почках.
Глюкозурия и, как следствие, полиурия развиваются при достижении концентрации глюкозы в крови 9,9 ммоль/л, когда преодолевается почечный порог, и глюкоза появляется в моче.
Гиперлактатацидемия развивается вследствие торможения катаболизма лактата в ЦТК и нарушения ресинтеза пирувата из лактата (см. рис. 12-17).
2. Нарушения липидного обмена при отсутствии инсулина характеризуются усилением липолиза и снижением липогенеза. Дефицит инсулина при сахарном диабете приводит к тому, что:
а) контринсулярные гормоны (глюкагон, адреналин, глюкокортикоиды и др.) стимулируют мобилизацию липидов из жировых депо и доставку жирных кислот к органам, что является адаптивным механизмом, поставляющим альтернативный субстрат окисления в условиях снижения утилизации глюкозы клетками;
б) снижается активность ЛП-липазы адипоцитов, поэтому свободные жирные кислоты не поступают в жировую ткань;
в) начинает преобладать эффект глюкагона, стимулирующий кетогенез в печени и гормончувствительную ТАГ-липазу в адипоцитах;
г) в норме кетоновые тела стимулируют выход инсулина из поджелудочной железы, что угнетает липолиз и таким образом ограничивает доставку липидов в печень и соответственно кетогенез (при сахарном диабете этот регуляторный механизм нарушен: идет усиленная продукция кетоновых тел печенью благодаря интенсивному β-окислению жирных кислот);
д) при сахарном диабете в избыточном количестве образуется продукт β-окисления жирных кислот — ацетил-КоА, однако способность цикла Кребса утилизировать данный продукт существенно снижена. В результате этого избыток ацетил-КоА становится источником образования больших количеств кетоновых тел: β-оксимасляной, ацетоуксусной кислот и ацетона, что и приводит к кетонемии. Кетоновые тела начинают выделяться с мочой в виде натриевых солей (кетонурия), а ацетон — также и в составе выдыхаемого воздуха.
3. Нарушения белкового обмена при отсутствии инсулина характеризуются преобладанием процессов катаболизма вследствие активации глюконеогенеза из глюкогенных аминокислот и снижения проницаемости клеточных мембран для аминокислот, что приводит к недостатку в тканях свободных аминокислот и нарушению процесса синтеза белка. Стимулируется синтез мочевины, что характеризуется гиперазотемией и приводит к отрицательному азотистому балансу.
4. Нарушения кислотно-щелочного баланса организма развиваются в связи с накоплением кислых продуктов метаболизма в результате переключения аэробных путей утилизации глюкозы на анаэробный гликолиз с повышенной продукцией лактата и усиления ке-
тогенеза. По мере истощения емкости буферных систем организма формируется декомпенсированный метаболический ацидоз.
5. Водно-солевой обмен и дегидратация клеток. Нарушения водного обмена при СД проявляются полиурией (суточный диурез достигает 4-10 л) и полидипсией (до 9 л воды в сутки), обусловленной гипогидратацией организма и гиперосмией крови в связи с гипергликемией, гиперазотемией, кетонемией, гиперлактатацидемией, повышением содержания отдельных ионов. Именно эти явления дали основания для первоначального названия заболевания: diabetes mellitus (лат.) — сахарное мочеизнурение. Повышение осмотического давления крови сопровождается дегидратацией клеток, особенно чувствительны к этому клетки нервной ткани.
Необходимо особо отметить, что сочетание ацидоза и явлений дегидратации в эритроцитах приводит к снижению в этих клетках концентрации 2,3-дифосфоглицериновой кислоты — аллостерического модулятора функций гемоглобина. В этих условиях сродство гемоглобина к кислороду возрастает, но его способность отдавать кислород тканям уменьшается, вследствие чего гипоксия, вызванная глубокими метаболическими нарушениями при СД, усугубляется.
В результате перечисленных выше нарушений обмена веществ при СД могут развиваться серьезные осложнения, к которым относят комы и диабетические микро- и макроангиопатии (нефропатия, слепота, синдром «диабетической стопы»), вторичные инфекции из-за иммунодефицита.
Диабетическая кома. Критическая дегидратация тканей организма с поражением функций головного мозга ведет к развитию диабетической (гипергликемической) комы. Кома развивается при достижении концентрации глюкозы в крови от 19,4 до 33,3 ммоль/л и более. В этих условиях вследствие кетоацидоза ионы калия выходят во внеклеточное пространство (гиперкалиемия), что лежит в основе нарушения сократительной функции миокарда, а также дыхательной мускулатуры. Диабетическая кома может привести к летальному исходу, если больному не будет своевременно проведена специфическая противокоматозная терапия.
Различают следующие виды диабетической комы:
1. Гипергликемическая кетоацидотическая кома. Развивается чаще всего у больных СД 1 типа вследствие гипергликемии, гиперкетонемии и метаболического ацидоза. Глюкоза и кетоновые тела выводятся с мочой (глюкозурия и кетонурия), что способ-
ствует увеличению осмотического давления в первичной моче, потере ионов Na и сопровождается полиурией. При этом возникает обезвоживание, которое ведет к недостаточности периферического кровообращения и гипоксии тканей. Ацидоз вызывает дыхание Куссмауля, при котором теряется СО2и как следствие усугубляются нарушения водно-электролитного баланса, кислотно-щелочного равновесия, возникает резкое нарушение метаболизма и функций клеток ЦНС, что приводит к расстройству высшей нервной деятельности. К клиническим проявлениям комы относятся: слабость, головная боль, адинамия, диспепсические расстройства (в 30-50% случаев — «абдоминальный синдром» — клиника «острого живота»), дыхание Куссмауля с запахом ацетона в выдыхаемом воздухе, снижение кровяного давления и частый слабый пульс, нарастающая сухость кожи и слизистых оболочек. Затем наступает полная потеря сознания, расслабление мышц, зрачки сужаются, отмечаются характерные признаки энцефалопатии. Содержание глюкозы в крови превышает 22 ммоль/л, кетоновых тел — 17 ммоль/л, повышено содержание остаточного азота, мочевины, холестерина, жирных кислот, уровень натрия чаще нормальный, реже — снижен, уровень калия чаще нормальный, у больных с почечной недостаточностью может быть повышен.
2. Гипергликемическая гиперосмолярная кома. Встречается реже, чем кетоацидотическая, и развивается у больных СД 2 типа старше 50 лет при дополнительном воздействии обезвоживающих факторов (рвота, понос, ограничение приема жидкости, ожоги, кровопотеря, полиурия, прием диуретиков). Основными звеньями патогенеза этого вида комы являются дегидратация организма и развитие гиперосмолярности плазмы, уровень гликемии может достигать 55 ммоль/л. У больных нет выраженной гиперкетонемии и кетонурии, отсутствует запах ацетона изо рта и, если не обратиться к врачу, нарастает уровень глюкозы в крови до крайне высокой степени, что способствует усилению диуреза (глюкозурический осмотический диурез). Возникающее обезвоживание приводит к гиповолемии, стимуляции секреции альдостерона и задержке ионов Na и Cl. Показатель осмолярности плазмы повышается в 1,5-2 раза (в норме около 300 мосмоль/л, при коме достигает 500 мосмоль/л), что приводит к резко выраженной внутриклеточной дегидратации, нарушению водного и электролитного равновесия в клетках мозга, гипоксии ЦНС с выраженной неврологической симптоматикой и потере сознания.
3. Гипергликемическая кома с лактат-ацидозом (лактацидотическая). Это относительно редкое, но опасное осложнение СД. В механизме ее развития важную роль играют следующие факторы:
а) снижение активности ферментативного пируватдегидрогеназного комплекса (выявляется при дефиците инсулина), превращающего пируват в ацетил-КоА. Пируват в обратимой реакции, катализируемой лактатдегдрогеназой, превращается в молочную кислоту (см. рис. 12-17);
б) применение лекарственных препаратов, стимулирующих анаэробный гликолиз и тем самым повышающих содержание лактата и пирувата в организме (например, бигуаниды, повышающие утилизацию глюкозы за счет ее анаэробного распада). При поражении печени или почек может иметь место кумуляция этих препаратов в организме, в результате чего развиваются лактоацидоз и кома;
в) гипоксическое состояние (при котором, как правило, стимулируется гликолиз), вызванное физическим переутомлением, сердечной или дыхательной недостаточностью.
Как следствие в крови накапливается молочная кислота (содержание лактата в плазме превышает 5 ммоль/л), что сопровождается развитием коллапса, нарушением сердечной деятельности и функций дыхательного центра (возникает патологическое дыхание Куссмауля), угнетением сознания, нарушением чувствительности, дисфункцией желудочно-кишечного тракта, резко выраженной дегидратацией тканей. Гиперкетонемия и кетонурия отсутствуют, могут выявляться незначительная гипергликемия и небольшая глюкозурия. Вследствие несвоевременной диагностики и трудности лечения прогноз может быть неблагоприятным.
4. Гипогликемическая кома. Связана с передозировкой инсулина, препаратов сульфонилмочевины, развитием вторичного гипопитуитаризма (следствие ангиопатии сосудов гипофиза), ослабляющего ответ на гипогликемию, и явлениями диабетического нефросклероза, что удлиняет время циркуляции инсулина и, кроме того, еще более снижает почечный порог для глюкозы, способствуя ее потере.
Причинами гипогликемии могут быть также гиперпродукция инсулина опухолью поджелудочной железы (инсулиномой), недостаточность контринсулярных гормонов, печеночные формы гликогенозов, заболевания печени, голодание, нарушение расщепления и всасывания углеводов в желудочно-кишечном тракте и др.
В механизме развития гипогликемической комы решающее значение имеет снижение доставки глюкозы к нервным клеткам, что ведет к их энергетическому истощению и нарушению функций ЦНС. При снижении уровня глюкозы менее 3 ммоль/л возникают потливость, тремор, чувство тревоги и голода, слабость. Затем развивается состояние, напоминающее алкогольное опьянение и сопровождающееся дезориентацией, агрессивностью, галлюцинациями. При дальнейшем падении содержания глюкозы (менее 2,5 ммоль/л) возникают клонические судороги и потеря сознания. В тяжелых случаях могут наступать отек и некроз отдельных участков мозга.
Микро- и макроангиопатии относят к более поздним осложнениям, развивающимся при СД обоих типов.
1. Микроангиопатия. Это осложнение выражается в повреждении сосудов микроциркуляции и развивается вследствие метаболических нарушений в сосудистой стенке (гликозилирование белков) и развития васкулита. Чаще всего поражаются сосуды почек и сетчатки глаза.
Поражение почек — диабетическая нефропатия из-за развития макро- и микроангиопатии в настоящее время является основной причиной ранней смертности у больных диабетом молодого возраста. При этом происходит избыточное гликозилирование коллагена базальных мембран почечных клубочков, приводящее к существенным нарушениям структуры и функций почечного фильтра.
Если в суточной моче концентрация альбумина превышает 30 мг (микроальбуминурия), и эти значения повторяются несколько раз, то необходимо проводить лечение, так как данные изменения характерны для начинающейся диабетической нефропатии. По мере прогрессирования поражения почек при диабете развивается выраженная протеинурия. Тщательный контроль за уровнем глюкозы в крови и лечение любых форм гипертонии могут приостановить микроальбуминурию и предупредить развитие манифестной почечной недостаточности.
Поражение сетчатки глаз при диабете — диабетическая ретинопатия относится к числу одной из наиболее частых причин развивающейся слепоты при этой патологии. Основную роль в патогенезе развития отечно-геморрагических форм диабетической ретинопатии играют максимальные суточные колебания уровня глюкозы в крови. Так, при повышении уровня глюкозы на 5,55
ммоль/л осмолярность сыворотки повышается на 5,5 мосмоль/л, что соответствует увеличению давления на 86 мм рт.ст., поэтому если увеличение концентрации сахара в крови идет быстро (в постабсорбтивном периоде), то в капиллярах сетчатки резко возрастает трансмуральное (разница между давлением внутри и снаружи сосуда) осмотическое давление. Жидкость «вытягивается» из ткани сетчатки в кровь, что ведет к резкому повышению трансмурального гидростатического давления в сосудах сетчатки. Повышение гидростатического давления за счет миогенной ауторегуляции приводит к сужению артериол вплоть до полного перекрытия кровотока (с развитием микроинфарктов сетчатки), а капилляры растягиваются с образованием микроаневризм и развития кровоизлияний.
Другой причиной, приводящей к потере зрения, является диабетическая катаракта, в патогенезе которой важную роль играет длительно существующая гипергликемия. Она вызывает, с одной стороны, усиление синтеза сорбитола и маннитола (ферментативное гликозилирование) и накопление этих углеводов в хрусталике глаза, приводящее по осмотическому градиенту к увеличению содержания в нем воды. С другой стороны, гликозилирование белков хрусталика и его капсулы (неферментативное) также вызывает необратимые нарушения его структуры.
2. Макроангиопатия. Диабетическая макроангиопатия проявляется атеросклерозом вследствие выраженного нарушения обмена липопротеинов. Особенностями атеросклероза при СД являются большая распространенность (захват многих сосудистых бассейнов), быстрое прогрессирование, начало в более молодом возрасте. Патологический процесс охватывает сосуды головного мозга, сердца, почек, а также сосуды конечностей, в особенности сосуды голени и стопы. Диабет, даже в условиях его лечения современными средствами, характеризуется ускоренными темпами старения организма. Наличием диабета обусловлены высокая частота инфарктов миокарда, инсультов и случаев гангрены пальцев ног или стопы.
В настоящее время считают, что диабет ускоряет развитие атеросклероза в результате:
• дислипопротенемии (см. раздел 12.5.2);
• гиперхолестеринемии (см. раздел 12.5.8);
• эндотелиальной дисфункции, возникающей из-за окислительного стресса, основным фактором которого являются модифицированные ЛПНП;
• активации неспецифического звена системы иммунитета, сопровождающейся выработкой провоспалительных цитокинов;
• эффекта гормона роста вследствие отсутствия противодействия со стороны инсулина в условиях его абсолютного или относительного дефицита, приводящего к усилению процесса пролиферации гладкомышечных клеток;
Диабетическая нейропатия — нарушение функции нервов — развивается вследствие ангиопатии из-за нарушения кровоснабжения нервов, гликозилирования белков нейронов и миелиновых оболочек, гиперосмолярного поражения шванновских клеток (проявление гиперосмотической дегидратации при СД). Эти нарушения способны вызывать дисфункции любой системы организма, имитируя многочисленные неврологические заболевания. В патологический процесс могут быть вовлечены как чувствительные, так и двигательные или вегетативные нервные волокна. В качестве типичных примеров клинического проявления диабетических нейропатий можно назвать образование трофических язв на стопах — «диабетическую стопу», различные расстройства функций желудочно-кишечного тракта, мочевого пузыря, импотенцию. При исследовании структуры нервных волокон диабетиков с помощью световой микроскопии часто выявляются их демиелинизация, истончение и склероз эпиневрия, отек и дистрофии нервных волокон, глиальная клеточная реакция.
Патогенез диабетических нейропатий полностью не раскрыт, однако в настоящее время можно назвать ряд факторов, безусловно определяющих развитие этого осложнения диабета:
1) нарушение структуры миелина, когда патологический процесс обусловливает изменение как химической структуры, так и количественного соотношения между основными биохимическими компонентами (холестерин, ТАГ, фосфолипиды, гликолипиды и белки) миелина нервных волокон. Клинические наблюдения свидетельствуют о том, что под влиянием заместительной терапии инсулином происходит существенная коррекция многих из указанных сдвигов;
2) сорбитоловый путь окисления глюкозы — в результате данного процесса происходит ферментативное окисление глюкозы с образованием сначала сорбитола, а затем фруктозы, повышенное количество которых приводит к гиперосмотической дегидратации нейронов;
3) токсическое действие пирувата.
Следует отметить, что общая черта всех вышеперечисленных нарушений, по крайней мере до развития глубоких морфологических изменений, — их обратимость под влиянием терапии, приводящей к снижению гипергликемии.
Нарушения углеводного обмена
При нарушении углеводного обмена могут развиваться состояния гипергликемии (концентрация глюкозы в крови более
 Рис. 12-21. Эффекты инсулина и глюкагона в норме и при инсулиновой недостаточности. В норме печень образует примерно 10 г/ч глюкозы, из них 65-75% в результате действия глюкагона на гликогенолиз, глюконеогенез. Инсулин может угнетать секрецию глюкагона α-клетками независимо от уровня глюкозы в крови. В печени инсулин угнетает образование глюкозы и кетоновых тел. При уменьшении отношения инсулин/глюка- гон увеличивается образование глюкозы и кетоновых тел в печени
Рис. 12-21. Эффекты инсулина и глюкагона в норме и при инсулиновой недостаточности. В норме печень образует примерно 10 г/ч глюкозы, из них 65-75% в результате действия глюкагона на гликогенолиз, глюконеогенез. Инсулин может угнетать секрецию глюкагона α-клетками независимо от уровня глюкозы в крови. В печени инсулин угнетает образование глюкозы и кетоновых тел. При уменьшении отношения инсулин/глюка- гон увеличивается образование глюкозы и кетоновых тел в печени
5,5 ммоль/л) и гипогликемии (менее 3,3 ммоль/л). Уровень глюкозы в крови зависит от вида пробы (цельная кровь, венозная или капиллярная, плазма) и от режима взятия пробы (натощак — уровень глюкозы утром после не менее чем 8-часового голодания; через 2 ч после глюкозотолерантного теста) (табл. 12-3). В клинике выделяют еще один показатель уровня глюкозы в крови — постпрандиальная гликемия — это уровень глюкозы через 2 ч после обычного приема пищи (не более 7,5 ммоль/л).
Таблица 12-3. Концентрация глюкозы в крови в норме (ммоль/л)
 Патогенез гипогликемий может быть связан с недостаточным поступлением глюкозы в кровь, ускоренным выведением ее из крови либо комбинацией этих факторов. Различают физиологическую и патологическую гипогликемию.
Патогенез гипогликемий может быть связан с недостаточным поступлением глюкозы в кровь, ускоренным выведением ее из крови либо комбинацией этих факторов. Различают физиологическую и патологическую гипогликемию.
Физиологическая гипогликемия. Выявляется при тяжелой и длительной физической нагрузке; длительном умственном напряжении; у женщин в период лактации; развивается сразу вслед за алиментарной гипергликемией благодаря компенсаторному выбросу в кровь инсулина.
Патологическая гипогликемия (гиперинсулинизм). Чаще возникает у больных сахарным диабетом в связи с передозировкой инсулина при лечении. Причиной ее могут быть также: аденома островковых клеток поджелудочной железы (инсулома); синдром Золлингера- Эллисона (аденома или карцинома поджелудочной железы, которая, по-видимому, развивается из α-клеток островков Лангерганса, ответственных за выделение глюкагона и гастрина).
Патологическая гипогликемия (без гиперинсулинизма). Выявляется: при патологии почек, сопровождающейся снижением порога для глюкозы, что приводит к потере глюкозы с мочой; нарушении всасывания углеводов; заболеваниях печени, сопровождающихся торможением синтеза гликогена и глюконеогенеза (острые и хронические гепатиты); недостаточности надпочечников (дефицит глюкокортикоидов); гипоавитаминозе В1, галактоземиях и при печеночных формах гликогенозов; голодании или недостаточном питании (алиментарная гипогликемия); недостаточности механизмов регуляции углеводного обмена у новорожденных.
Гипергликемия у человека встречается чаще, чем гипогликемия. Различают следующие типы гипергликемий.
Физиологические гипергликемии. Это быстрообратимые состояния. Нормализация уровня глюкозы в крови происходит без каких-либо внешних корригирующих воздействий. К ним относятся:
1. Алиментарная гипергликемия. Обусловлена приемом пищи, содержащей углеводы. Концентрация глюкозы в крови нарастает вследствие ее быстрого всасывания из кишечника. Активация секреции гормона β-клетками островков Лангерганса поджелудочной железы начинается рефлекторно, сразу после попадания пищи в полость рта и достигает максимума при продвижении пищи в двенадцатиперстную кишку и тонкий кишечник. Пики концентраций инсулина и глюкозы в крови совпадают по времени. Таким образом, инсулин не только обеспечивает доступность углеводов пищи к клеткам организма, но и ограничивает повышение концентрации глюкозы в крови, не допуская потерю ее с мочой.
2. Нейрогенная гипергликемия. Развивается в ответ на эмоциональный стресс и обусловлена выбросом в кровь большого количества катехоламинов, образующихся в мозговом веществе надпочечников и реализующих свои гипергликемические эффекты (см. табл. 12-2). Освобождающаяся глюкоза быстро выходит в кровь, обусловливая гипергликемию. Физиологический смысл этого феномена состоит в обеспечении срочной мобилизации резерва углеводов для использования их в качестве источников энергии (окисления) в предстоящей повышенной двигательной активности в условиях стресса.
Патологические гипергликемии. Причинами их развития являются:
1) нейроэндокринные расстройства, когда нарушены соотношения уровня гормонов гипо- и гипергликемического действия. Например, при заболеваниях гипофиза, опухолях коры надпочечников, при феохромоцитоме, гиперфункции щитовидной железы; при недостаточной продукции инсулина, глюкагономе;
2) органические поражения центральной нервной системы, расстройства мозгового кровообращения различной этиологии;
3) нарушения функций печени при циррозе;
4) судорожные состояния, когда происходит расщепление гликогена мышц и образование лактата, из которого в печени синтезируется глюкоза;
5) действие наркотических веществ (морфин, эфир), возбуждающих симпатическую нервную систему и тем самым способствующих развитию гипергликемии.
Наиболее часто встречается гипергликемия при недостаточности инсулина и (или) его действия, которая лежит в основе сахарного диабета.
Диагностика нарушений углеводного обмена методом нагрузок
Исследование состояния углеводного обмена с диагностической целью в клинике начинают с определения натощак содержания глюкозы в крови и анализа мочи на присутствие в ней глюкозы и кетоновых тел. Если результаты анализов свидетельствуют о наличии гипергликемии, глюкозурии и кетонурии, то этого оказывается достаточно для подтверждения диагноза сахарного диабета. Доказано, что ни одно из других заболеваний внутренних органов не дает всей триады: гипергликемии, глюкозо- и кетонурии. Лишь при сахарном диабете имеют место существенные нарушения не только углеводного, но и жирового обмена.
Для углубленного исследования состояния углеводного обмена используют методы «сахарных» нагрузок.
Проба с однократной сахарной нагрузкой для получения гликемической («сахарной») кривой. У обследуемого утром натощак берут кровь из пальца для определения концентрации глюкозы, после чего дают сахарную нагрузку: прием внутрь 100 г глюкозы, растворенной в 200 мл кипяченой воды. Время, в течение которого раствор следует выпить, не должно превышать 5 мин. Повторные заборы проб крови из пальца ведут с интервалом в 30 или 60 мин. Длительность пробы (у взрослых) составляет 3 ч. На основании полученных данных строят кривую, откладывая по оси ординат концентрацию глюкозы, а по оси абсцисс — время. Типы гликемических кривых, присущих норме или сахарному диабету, представлены на рис. 12-22.
Для гликемической кривой у здоровых субъектов характерны следующие признаки. Уже через 15 мин после приема раствора
 Рис. 12-22. Типы гликемических кривых в норме и в состояниях, характеризующихся пониженной толерантностью к глюкозе: 1 — норма; 2 — латентный сахарный диабет (легкая форма заболевания); 3 — тяжелая форма сахарного диабета
Рис. 12-22. Типы гликемических кривых в норме и в состояниях, характеризующихся пониженной толерантностью к глюкозе: 1 — норма; 2 — латентный сахарный диабет (легкая форма заболевания); 3 — тяжелая форма сахарного диабета
глюкозы внутрь в крови начинает расти концентрация глюкозы, достигая максимума к концу первого часа (в промежутке от 30-й до 60-й мин). При этом концентрация глюкозы превышает таковую натощак на 50-75%. Далее концентрация глюкозы в крови начинает снижаться, и к окончанию второго часа наблюдения она либо достигает исходного уровня (натощак), либо падает ниже исходного уровня (вариант физиологической гипогликемии), либо остается несколько повышенной, но не превышает 6,6 ммоль/л. К третьему часу во всех трех возможных вариантах концентрация глюкозы в крови не отличается от исходного значения (натощак).
У больных сахарным диабетом концентрация глюкозы натощак повышена, а нарастание гликемической кривой после сахарной нагрузки происходит медленнее. Максимальное значение показателя регистрируют только через 60-150 мин от начала наблюдения, при этом концентрация глюкозы может в 1,8 раза превышать ее исходное значение. Спад концентрации глюкозы крови (гипогликемическая фаза) также происходит чрезвычайно медленно (вплоть до отсутствия такового), что коррелирует со степенью тяжести заболевания. Если же понижение концентрации глюкозы все же происходит, то оно растягивается на 3-4 ч.
Тест толерантности к глюкозе (ГТТ). Кровь для проведения теста также берут из пальца дважды: натощак и спустя 120 мин после нагрузки глюкозой. Согласно критериям ВОЗ, у практически здорового человека концентрация глюкозы в крови натощак не должна превышать 5,5 ммоль/л. Спустя 120 мин после стандартной нагрузки глюкозой (одномоментный прием внутрь 75 г глюкозы в 300 мл воды) уровень глюкозы не должен превышать 6,7 ммоль/л. В случае если концентрация глюкозы в крови натощак превышает 6,1 ммоль/л, а тест свидетельствует о том, что спустя 120 мин уровень глюкозы остается выше 7,8 ммоль/л, то это веский аргумент в пользу если не диабета, то преддиабетического состояния у обследуемого.
Ожирение — избыточное отложение жира в жировой ткани. Ожирение значительно повышает риск развития артериальной гипертонии, сахарного диабета 2 типа, атеросклероза, поэтому очень важно следить за своим весом. Считается, что человек страдает ожирением, если его масса превышает нормальную более чем на 20% и продолжает увеличиваться далее. Этим недугом страдает более трети взрослого населения России. В 1998 г. Всемирная организация здравоохранения (ВОЗ) признала ожирение хроническим заболеванием. По статистике ВОЗ, в экономически развитых странах около 30% взрослых и до 10% детей имеют ту или иную форму и степень ожирения. В возрастных группах после 50 лет это заболевание встречается чаще. За последнее десятилетие число таких больных в мире увеличилось почти в два раза и по оценке специалистов в 2025 г. их количество составит 300 млн человек. Ситуация тем более сложная, что с каждым годом увеличивается число молодых людей, страдающих ожирением, снижается общая продолжительность жизни населения земного шара в связи с тяжелыми заболеваниями, сопутствующими ожирению (см. табл. 12-5).
У тучных людей старше 50 лет смертность увеличивается на 50% по сравнению с лицами, не имеющими ожирения. У женщин с ожирением чаще встречаются рак эндометрия, яичника, шейки матки, желчного пузыря и молочной железы, у мужчин — рак предстательной железы и толстой кишки, снижение потенции. Жировая ткань, составляющая в норме 15-20% от массы тела у мужчин и 20-29% у женщин, — это метаболически активное образование.
Таблица 12-5. Заболевания, сопутствующие ожирению (по С. Бутровой,
Адипоциты секретируют гемопоэтины; выделяют цитокины — TNF-α, IL-6, трансформирующий фактор роста (TGF) β и соответствующие им растворимые рецепторы; синтезируют биоактивные вещества — ангиотензиноген, ингибитор активатора плазминогена; ряд ферментов (липопротеиновая липаза, индуцируемая NO-синтаза, аполипопротеин Е) и гормонов (лептин, резистин, адипонектин, эстрогены); выделяют свободные жирные кислоты. Увеличение массы жировой ткани влечет за собой повышение содержания лептина в крови, причем его продукция в подкожной жировой клетчатке выше, чем в висцеральных жировых депо. Уровень лептина отражает не только количество накопленного жира, но и нарушения энергетического обмена: при голодании он значи-
тельно снижается, при переедании — повышается. Избыток лептина вызывает инсулинорезистентность скелетных мышц и жировой ткани и подавляет действие инсулина на клетки печени (инсулин активирует адипоциты, повышая образование лептина, в свою очередь, лептин, воздействуя на собственные рецепторы, локализованные на поверхности β-клеток, тормозит секрецию инсулина).
У женщин наличие достаточно выраженной жировой ткани существенно для поддержания нормальной половой функции. Менструации у девочек, не достигших критической массы (около 48 кг), не начинаются, даже если пубертатный период пройден. При похудении на 10-15% нормы даже при сохранении цикла нет овуляции, возможна и полная аменорея. Эти изменения обратимы, при нормализации веса детородная функция восстанавливается. Вероятно, невозможность деторождения у женщин, не имеющих достаточных жировых запасов для успешного рождения и вскармливания ребенка, была выработана в процессе естественного отбора. У мужчин похудение и физические нагрузки приводят к уменьшению полового влечения, а если вес тела на 25% ниже нормы, выработка спермы угнетается. Следует отметить, что сильное развитие мускулатуры (например, у культуристов) действует на репродуктивные процессы так же, как и похудение. Избыток эстрогенов у полных мужчин приводит к снижению потенции, гинекомастии, гипогонадизму с понижением уровня тестостерона.
Классификация ожирения. Ожирение может возникать как самостоятельное заболевание — в этом случае говорят о первичном ожирении. Вторичное ожирение — это синдром, возникающий вследствие гормональных или других расстройств в организме.
Первичное ожирение возникает при нарушении гормональной связи между жировой тканью и гипоталамусом. Это генетически опосредованное нейроэндокринное заболевание, его главная черта — абсолютная или относительная лептиновая недостаточность. Около 20% больных имеют абсолютную лептиновую недостаточность, однако дефицит лептина 1 не является основной причиной развития ожирения. Более 80% пациентов с первичным ожирени-
1 Генетически опосредованный дефицит лептина проявляется ранним ожирением, пониженным обменом веществ, гипогонадотропным гипогонадизмом, гиперинсулинемией, нарушением гипоталамо-питуитарных и тиреоидальных взаимодействий и нарушением количества и функции Т-лимфоцитов, что повышает восприимчивость больных к инфекциям. Известны 5 отдельных мутаций в гене лептина, вызывающих развитие первичного ожирения.
ем имеют выраженную гиперлептинемию, что свидетельствует о резистентности к гормону. Известны следующие механизмы резистентности:
• нарушение транспорта лептина через гематоэнцефалический барьер (введение гормона даже в больших дозах не дает результата);
• нарушение переноса гормона транспортными белками;
• мутации рецептора лептина 1 (несмотря на продукцию лептина, центр голода продолжает секрецию нейропептида Y);
• мутации генов, кодирующих рецепторы к меланокортину 2 (4% всех больных с ожирением). Следует отметить, что на фоне введения лептина у млекопитающих снижается только масса жировой ткани, в то время как при голодании снижается также масса других тканей.
Вторичное ожирение — синдром, возникающий при нарушении соотношения между процессами липолиза и липогенеза, носит симптоматический характер и порождается различными расстройствами (эндокринопатии, опухоли мозга, нарушения мозгового кровообращения и пр.).
По степени увеличения массы тела различают ожирение I степени (масса тела увеличена на 30%); II степени (на 30-50%); III степени (более чем на 50%).
Одним из наиболее распространенных показателей для оценки степени ожирения является индекс массы тела (ИМТ), рассчитываемый следующим образом:
Больные с ИМТ 30 кг/м 2 и более, а также пациенты с ИМТ 27 кг/м 2 или более, ожирение которых связано с такими факторами риска, как диабет 2 типа или дислипопротеинемия, подлежат обязательному лечению (табл. 12-6).
1 Дети с дефектом рецептора лептина быстро набирают избыточную массу в течение первых месяцев жизни, отличаются гиперфагией и агрессивным поведением во время еды. Иногда дефект рецептора имеет более выраженные проявления (гипотиреоз), чем отсутствие лиганда.
2 Меланокортины (адренокортикотропный и меланоцитостимулирующий гормоны, а также и их фрагменты) образуются в гипофизе из проопиомеланокортина. Лептин стимулирует экспрессию гена проопиомеланокортина, образующийся проопиомеланокортин расщепляется до субстрата, который действует как супрессор пищевого поведения, возможно через MC4R. При снижении гипоталамического меланокортинэргетического сигнала через рецепторы MC4R наблюдаются гиперфагия и прибавка массы.
Таблица 12-6. Классификация избыточной массы у взрослых в зависимости от индекса массы тела (в соответствии с докладом ВОЗ 1998 г.)
Наиболее простым методом определения склонности к ожирению является измерение окружности талии. В идеале окружность талии не должна превышать 94 см у мужчин и 80 см у женщин. Если окружность талии у мужчин достигает 102 см, а у женщин — 88 см, возникает серьезная угроза увеличения риска заболевания.
По особенностям морфологии жировой ткани выделяют гипертрофическое и гиперпластическое ожирение.
Гипертрофическое ожирение связано с увеличением размеров адипоцитов (это лабильный фактор, зависимый от питания), чаще встречается в зрелом возрасте. При этом виде ожирения масса тела может увеличиваться в 3-3,3 раза.
Гиперпластическое ожирение сопровождается увеличением количества адипоцитов. Начинается, как правило, в детском возрасте, так как дифференцировка фибробластических клеток-предшественниц в новые адипоциты во взрослом организме — явление довольно редкое (это происходит в период внутриутробного развития и в раннем грудном возрасте). В развитии гиперпластического ожирения огромное значение имеет наследственность, определяющая пролиферативные возможности этих клеток. Избыток массы тела при гиперпластическом ожирении может достигать гигантских величин (до 1000%). Следует отметить, что в подростковый и предклимактерический периоды повышается пролиферативная активность преадипоцитов. Кроме того, их деление индуцируют избыточная калорийность пищи, сахарный диабет или переедание у беременных. В этих случаях гиперпластическое ожирение развивается у взрослых.
Жир может располагаться в подкожно-жировой клетчатке (подкожный жир) и вокруг внутренних органов (висцеральный жир), вместе подкожно-жировая клетчатка в области живота и висцеральный жир брюшной полости составляют абдоминальный жир. Разная локализация жировых отложений при различных формах первичного и вторичного ожирения зависит от влияния мужских и женских половых гормонов на распределение и катехоламиновых рецепторов в разных отсеках жировой ткани. Жировая ткань, локализованная в различных частях тела, отличается по своей гормональной функции (см. раздел 12.4). У людей, склонных к первичному ожирению, уменьшена экспрессия β-адренорецепторов на адипоцитах.
В зависимости от характера распределения жировой ткани различают:
ожирения, когда избыточные отложения жира располагаются на животе и верхней части туловища (наиболее характерен для мужчин) (рис. 12-34);
• гиноидный (грушевидный) тип ожирения, когда избыточные отложения жира располагаются на бедрах, ягодицах и в нижней части туловища (наиболее характерен для женщин);
• смешанный тип ожирения — комбинирует признаки андроидного и гиноидного типов.
Гиноидное ожирение чаще носит гиперпластический характер, поэтому оно более резистентно к диетотерапии. Однако более патогенным считается андроидное, а более благоприятными — гиноидное, смешанное.
Отложение жировой клетчатки в абдоминальной области (яблочный или верхний тип ожирения) больше связано с заболеваемо-
стью и смертностью, чем гиноидный или нижний тип ожирения, и даже больше, чем степень ожирения. Большое количество абдоминального жира способствует развитию дислипидемии, сахарного диабета, сердечно-сосудистых заболеваний, у женщин — возникновению саркомы. Эта зависимость не связана с общим содержанием жира в организме. При одинаковом ИМТ абдоминальное ожирение имеет более высокий риск развития сопутствующих заболеваний, чем ожирение по нижнему типу, что увеличивает смертность у людей.
По этиологии ожирение классифицируют на экзогенно-конституциональное, гипоталамическое, гормональное (эндокринное).
Экзогенно-конституциональное ожирение (часто, но не всегда относится к первичной форме ожирения). Нарушение пищевого поведения (например, синдром ночной еды, повышенное потребление пищи в ответ на стресс) приводит к отложению избытка жира в организме в соответствии с формулой:
Отложение жира = Поступление энергии — Расход энергии.
Длительное повышение активности «пищевого центра» ведет к повышению аппетита (гиперфагии) и ожирению (рис. 12-35). Привычка переедать может быть приобретена в детстве. Так, установлено, что избыточное кормление ребенка первого года жизниспособствует развитию гиперпластического ожирения, характеризующегося увеличением объема жировых клеток.
Гипоталамическое ожирение. Является следствием поражения области гипоталамуса. Причиной могут быть перенесенные травмы головного мозга, стойкая внутричерепная гипертензия, опухоли мозга, менингит, а также врожденные дегенеративные изменения гипоталамической области (например, синдром Фрелиха) (рис. 12-36).
Гормональное ожирение. Связано как с гипо-, так и с гиперфункцией желез внутренней секреции и развивается при гипотиреозе, гипофункции половых желез, а также при гиперинсулинизме и гиперкортицизме. В крови таких больных повышается содержание ЛПНП и ЛПОНП, НЭЖК. При гормональном ожирении рано развивается гипертриацилглицеролемия и несколько позже — гиперхолестеринемия. Нарушению обмена липидов сопутствует изменение углеводного обмена: развивается гипергликемия, стимулирующая секрецию инсулина и его предшественника. В свою очередь, секрецию проинсулина и инсулина стимулируют НЭЖК, ЛПОНП, ЛПНП. Усиленный выброс глюкокортикоидов, стимулирующих глюконеогенез, также повышает уровень инсулина в крови.
По патогенезу различают алиментарное, метаболическое и энергетическое ожирение.
Алиментарное ожирение — развивается при чрезмерном потреблении пищи, что может быть обусловлено:
а) нарушением деятельности гипоталамического пищевого центра (абсолютная или относительная лептиновая недостаточность, длительное возбуждение вентролатеральных ядер в результате травм, кровоизлияний, воспаления в диэнцефальной области (по этиологии это экзогенно-конституциональное или гипоталамическое ожирение);
б) афферентной импульсацией при частом возбуждении вкусовых рецепторов;
в) переходом от активного к малоподвижному образу жизни. При этом в некоторых случаях сохраняется высокий уровень возбудимости пищевого центра (характерный для лиц физического труда или спортсменов), что приводит к систематическому перееданию;
г) чрезмерным растяжением стенок желудка при его переполнении. Это снижает чувствительность нервных окончаний слизистой оболочки, и тормозящие импульсы передаются в пищевой центр только при очень большом скоплении пищи в желудке. В результате переедание становится постоянным и возникает ожирение;
д) пожилым возрастом, что объясняется несоответствием между прежним уровнем возбудимости центра голода и меньшими энергозатратами (после 25 лет основной обмен снижается в каждые последующие 10 лет примерно на 7,5%). Интересно отметить, что в глубокой старости часто развивается исхудание, поскольку угнетается активность пищевого центра и снижается переход углеводов в жиры.
Метаболическое ожирение обусловлено повышенным синтезом жира из углеводов. В обычных условиях до 30% поступающей в организм глюкозы под действием инсулина превращается в жир. При гиперфункции инсулярного аппарата этот процент возрастает. Аналогичное изменение метаболизма развивается при повышенной продукции пролактина (гормона передней доли гипофиза), глюкокортикоидов (по этиологии это гормональное ожирение).
Энергетическое ожирение обусловлено недостаточным использованием жиров в качестве источника энергии. Развивается при гиподинамии в сочетании с хорошим аппетитом, при снижении тонуса симпатической нервной системы и недостаточной продукции жиромобилизующих гормонов (СТГ, тиреоидные гормоны, катехоламины), поскольку задерживается выход жира из депо и использование его в качестве энергетического субстрата (по этиологии соответствует экзогенно-конституциональному или гормональному ожирению).
Последствия ожирения. При ожирении постепенно изменяется белковый обмен, который характеризуется снижением уровня общего белка крови преимущественно за счет уменьшения концентрации альбуминов, увеличением содержания фибриногена, продуктов деградации фибрина, снижением уровня гепарина. Следствием этого является нарушение транспорта НЭЖК и других липидов, снижение фибринолитической активности и повышение тромбогенных свойств крови, возникновение тромбоэмболических осложнений. Эти изменения являются факторами риска атеросклероза, ишемической болезни сердца, инсульта, гипертонической болезни. Возникают нарушения функций ЦНС: отмечаются утомляемость, сонливость, ухудшение памяти; развивается преждевременное старение, возникают изменения во внутренних
 Рис. 12-37.
Рис. 12-37.
Причины жировой инфильтрации печени: 1 — усиленный выход неэтерифицированных жирных кислот из жировой ткани; 2 — интенсивное длительное поступление хиломикронов из кишечника в кровь, а затем в печень; 3 — задержка окисления жирных кислот в печени до кетоновых тел; 4 — задержка выхода из печени пре-β- и β-липопротеинов. Затемненные прямоугольники — места нарушения процесса
органах, например жировая инфильтрация (ожирение или жировая трансформация) печени (рис. 12-37).
При первичном ожирении многие из расстройств метаболизма после нормализации веса корректируются (уменьшается или совсем проходит инсулинорезистентность, гипер- и дислипопротеинемия). Тем не менее у больного сохраняется лептиновая недостаточность, повышена активность липопротеиновой липазы жировой ткани, снижена реакция центров насыщения на серотонин, а адипоцитов — на β-адреномиметики, нарушена рецепция инсулина в гипоталамусе, а при гиперпластическом и смешанном ожирении увеличено число адипоцитов и т.д.
При быстрой нормализации веса снижается продукция тиреотропина, ухудшается холодовая адаптация. При дальнейшем падении массы тела еще больше снижается основной обмен. Отмечается тенденция к лейкопении, брадикардии и гипотонии, снижается иммунитет. У женщин возможно нарушение овариально-менструального цикла, которое связано со снижением эстрогенпродуцирующей функции адипоцитов. Многие похудевшие пациенты испытывают дисфорию, отмечаются обсессивные неврозы в связи с понижением выработки опиатных пептидов. Не-
которые психосоматические особенности похудевших пациентов с первичным ожирением напоминают таковые при психогенной анорексии. При голодании, диетах отмечается недостаток выделения серотонина, норадреналина, β-эндорфина и других биологически активных веществ в кровь. Снижение уровня серотонина субъективно воспринимается организмом человека как состояние депрессии, уменьшение концентрации норадреналина — упадка сил, β-эндорфина — неудовольствия, дискомфорта. Напротив, выделение норадреналина после еды вызывает чувство прилива сил, энергии, увеличивает уровень основного обмена. У людей с нарушениями центральной серотонинергической системы особенно сильны негативные реакции на голод, выражающиеся в снижении продукции серотонина. Даже при незначительном голодании у них развивается выраженная депрессия.
Адекватное лечение больного ожирением возможно лишь под наблюдением врача и не должно быть только симптоматическим, т. е. сводиться к диетотерапии и лечебной гимнастике. После открытия лептина большие надежды связывали с его применением для лечения лептиновой недостаточности при первичном ожирении.