75. Кетоновые тела, биосинтез и использование в качестве источников энергии. Причины развития кетонемии и кетонурии при голодании и сахарном диабете.
При голодании, длительной физической работе и в случаях, когда клетки не получают достаточного количества глюкозы, жирные кислоты используются многими тканями как основной источник энергии. В отличие от других тканей мозг и другие отделы нервной ткани практически не используют жирные кислоты в качестве источника энергии. В печени часть жирных кислот превращается в кетоновые тела, которые окисляются мозгом, нервной тканью, мышцами, обеспечивая достаточное количество энергии для синтеза АТФ и уменьшая потребление глюкозы. К кетоновым телам относят β-гидроксибутират, ацетоацетат и ацетон. Первые две молекулы могут окисляться в тканях, обеспечивая синтез АТФ. Ацетон образуется только при высоких концентрациях кетоновых тел в крови и, выделяясь с мочой, выдыхаемым воздухом и потом, позволяет организму избавляться от избытка кетоновых тел.
Синтез кетоновых тел в печени. При низком соотношении инсулин/глюкагон в крови в жировой ткани активируется распад жиров. Жирные кислоты поступают в печень в большем количестве, чем в норме, поэтому увеличивается скорость β-окисления. Скорость реакций ЦТК в этих условиях снижена, так как оксалоацетат используется для глюконеогенеза. В результате скорость образования ацетил-КоА превышает способность ЦТК окислять его. Ацетил-КоА накапливается в митохондриях печени и используется для синтеза кетоновых тел. Синтез кетоновых тел происходит только в митохондриях печени.
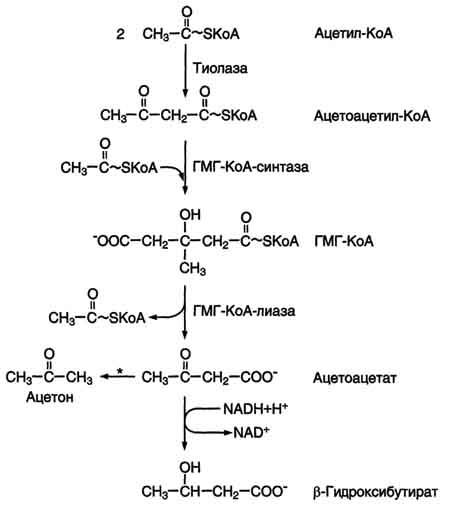

Рис. 8-33. Синтез кетоновых тел в митохондриях
гепатоцитов. Регуляторный фермент синтеза кетоновых тел (ГМГ-КоА-синтаза) ингибируется свободным КоА. — реакция идёт неферментативно при высокой концентрации кетоновых тел в крови.
Окисление кетоновых тел в тканях.
Кетоацидоз. В норме концентрация кетоновых тел в крови составляет 1-3 мг/дл (до 0,2 мМ/л), но при голодании значительно увеличивается. Увеличение концентрации кетоновых тел в крови называют кетонемией, выделение кетоновых тел с мочой — кетонурией. Накопление кетоновых тел в организме приводит к кетоацидозу: уменьшению щелочного резерва (компенсированному ацидозу), а в тяжёлых случаях — к сдвигу рН (некомпенсированному ацидозу), так как кетоновые тела (кроме ацетона) являются водорастворимыми органическими кислотами (рК
3,5), способными к диссоциации:
Ацидоз достигает опасных величин при сахарном диабете, так как концентрация кетоновых тел при этом заболевании может доходить до 400-500 мг/дл. Тяжёлая форма ацидоза — одна из основных причин смерти при сахарном диабете. Накопление протонов в крови нарушает связывание кислорода гемоглобином, влияет на ионизацию функциональных групп белков, нарушая их кон-формацию и функцию.
76. Холестерин. Пути поступления, использования и выведения из организма. Уровень холестерина в сыворотке крови. Биосинтез холестерина, его этапы. Регуляция синтеза.
Холестерол — стероид, характерный только для животных организмов. Он синтезируется во многих тканях человека, но основное место синтеза — печень. В печени синтезируется более 50% холестерола, в тонком кишечнике — 15- 20%, остальной холестерол синтезируется в коже, коре надпочечников, половых железах. В сутки в организме синтезируется около 1 г холестерола; с пищей поступает 300-500 мг (рис. 8-65). Холестерол выполняет много функций: входит в состав всех мембран клеток и влияет на их свойства, служит исходным субстратом в синтезе жёлчных кислот и стероидных гормонов. Предшественники в метаболическом пути синтеза холестерола превращаются также в убихинон — компонент дыхательной цепи и долихол, участвующий в синтезе гликопротеинов. Холестерол за счёт своей гидроксильной группы может образовывать эфиры с жирными кислотами. Этерифицированный холестерол преобладает в крови и запасается в небольших количествах в некоторых типах клеток, использующих его как субстрат для синтеза других веществ. Холестерол и его эфиры — гидрофобные молекулы, поэтому они транспортируются кровью только в составе разных типов ЛП. Обмен холестерола чрезвычайно сложен — только для его синтеза необходимо осуществление около 100 последовательных реакций. Всего в обмене холестерола участвует около 300 разных белков. Нарушения обмена холестерола приводят к одному из наиболее распространённых заболеваний — атеросклерозу. Смертность от последствий атеросклероза (инфаркт миокарда, инсульт) лидирует в общей структуре смертности населения. Атеросклероз — «полигенное заболевание», т.е. в его развитии участвуют многие факторы, важнейшие из которых наследственные. Накопление холестерола в организме приводит к развитию и другого распространённого заболевания — желчнокаменной болезни.
А. Синтез холестерола и его регуляция
Реакции синтеза холестерола происходят в цитозоле клеток. Это один из самых длинных метаболических путей в организме человека.
68. Кетоновые тела, биосинтез и использование в качестве источников энергии. Причины развития кетонемии и кетонурии при голодании и сахарном диабете.
При голодании, длительной физической работе и в случаях, когда клетки не получают достаточного количества глюкозы, жирные кислоты используются многими тканями как основной источник энергии. В отличие от других тканей мозг и другие отделы нервной ткани практически не используют жирные кислоты в качестве источника энергии. В печени часть жирных кислот превращается в кетоновые тела, которые окисляются мозгом, нервной тканью, мышцами, обеспечивая достаточное количество энергии для синтеза АТФ и уменьшая потребление глюкозы. К кетоновым телам относят β-гидроксибутират, ацетоацетат и ацетон. Первые две молекулы могут окисляться в тканях, обеспечивая синтез АТФ. Ацетон образуется только при высоких концентрациях кетоновых тел в крови и, выделяясь с мочой, выдыхаемым воздухом и потом, позволяет организму избавляться от избытка кетоновых тел.
Синтез кетоновых тел в печени. При низком соотношении инсулин/глюкагон в крови в жировой ткани активируется распад жиров. Жирные кислоты поступают в печень в большем количестве, чем в норме, поэтому увеличивается скорость β-окисления. Скорость реакций ЦТК в этих условиях снижена, так как оксалоацетат используется для глюконеогенеза. В результате скорость образования ацетил-КоА превышает способность ЦТК окислять его. Ацетил-КоА накапливается в митохондриях печени и используется для синтеза кетоновых тел. Синтез кетоновых тел происходит только в митохондриях печени.


Рис. 8-33. Синтез кетоновых тел в митохондриях
гепатоцитов. Регуляторный фермент синтеза кетоновых тел (ГМГ-КоА-синтаза) ингибируется свободным КоА. — реакция идёт неферментативно при высокой концентрации кетоновых тел в крови.
Окисление кетоновых тел в тканях.
Кетоацидоз. В норме концентрация кетоновых тел в крови составляет 1-3 мг/дл (до 0,2 мМ/л), но при голодании значительно увеличивается. Увеличение концентрации кетоновых тел в крови называют кетонемией, выделение кетоновых тел с мочой — кетонурией. Накопление кетоновых тел в организме приводит к кетоацидозу: уменьшению щелочного резерва (компенсированному ацидозу), а в тяжёлых случаях — к сдвигу рН (некомпенсированному ацидозу), так как кетоновые тела (кроме ацетона) являются водорастворимыми органическими кислотами (рК
3,5), способными к диссоциации:
Ацидоз достигает опасных величин при сахарном диабете, так как концентрация кетоновых тел при этом заболевании может доходить до 400-500 мг/дл. Тяжёлая форма ацидоза — одна из основных причин смерти при сахарном диабете. Накопление протонов в крови нарушает связывание кислорода гемоглобином, влияет на ионизацию функциональных групп белков, нарушая их кон-формацию и функцию.
69. Холестерин. Пути поступления, использования и выведения из организма. Уровень холестерина в сыворотке крови. Биосинтез холестерина, его этапы. Регуляция синтеза.
Холестерол — стероид, характерный только для животных организмов. Он синтезируется во многих тканях человека, но основное место синтеза — печень. В печени синтезируется более 50% холестерола, в тонком кишечнике — 15- 20%, остальной холестерол синтезируется в коже, коре надпочечников, половых железах. В сутки в организме синтезируется около 1 г холестерола; с пищей поступает 300-500 мг (рис. 8-65). Холестерол выполняет много функций: входит в состав всех мембран клеток и влияет на их свойства, служит исходным субстратом в синтезе жёлчных кислот и стероидных гормонов. Предшественники в метаболическом пути синтеза холестерола превращаются также в убихинон — компонент дыхательной цепи и долихол, участвующий в синтезе гликопротеинов. Холестерол за счёт своей гидроксильной группы может образовывать эфиры с жирными кислотами. Этерифицированный холестерол преобладает в крови и запасается в небольших количествах в некоторых типах клеток, использующих его как субстрат для синтеза других веществ. Холестерол и его эфиры — гидрофобные молекулы, поэтому они транспортируются кровью только в составе разных типов ЛП. Обмен холестерола чрезвычайно сложен — только для его синтеза необходимо осуществление около 100 последовательных реакций. Всего в обмене холестерола участвует около 300 разных белков. Нарушения обмена холестерола приводят к одному из наиболее распространённых заболеваний — атеросклерозу. Смертность от последствий атеросклероза (инфаркт миокарда, инсульт) лидирует в общей структуре смертности населения. Атеросклероз — «полигенное заболевание», т.е. в его развитии участвуют многие факторы, важнейшие из которых наследственные. Накопление холестерола в организме приводит к развитию и другого распространённого заболевания — желчнокаменной болезни.
А. Синтез холестерола и его регуляция
Реакции синтеза холестерола происходят в цитозоле клеток. Это один из самых длинных метаболических путей в организме человека.
55. Синтез и использование кетоновых тел. Гиперкетонемия, кетонурия, ацидоз при сахарном диабете и голодании.
Синтез кетоновых тел в печени. При низком соотношении инсулин/глюкагон в крови в жировой ткани активируется распад жиров. Жирные кислоты поступают в печень в большем количестве, чем в норме, поэтому увеличивается скорость β-окисления (рис. 8-32). Скорость реакций ЦТК в этих условиях снижена, так как оксалоацетат используется для глюконеогенеза. В результате скорость образования ацетил-КоА превышает способность ЦТК окислять его. Ацетил-КоА накапливается в митохондриях печени и используется для синтеза кетоновых тел. Синтез кетоновых тел происходит только в митохондриях печени.
Синтез кетоновых тел начинается с взаимодействия двух молекул ацетил-КоА, которые под действием фермента тиолазы образуют ацетоацетил-КоА (рис. 8-33). С ацетоацетил-КоА взаимодействует третья молекула ацетил-КоА, образуя 3-гидрокси-3-метилглутарил-КоА (ГМГ-КоА). Эту реакцию катализирует фермент ГМГ-КоА-синтаза. Далее ГМГ-КоА-лиаза катализирует расщепление ГМГ-КоА на свободный ацетоацетат и ацетил-КоА.
Ацетоацетат может выделяться в кровь или превращаться в печени в другое кетоновое тело — β-гидроксибутират путём восстановления.
В клетках печени при активном β-окислении создаётся высокая концентрация NADH. Это способствует превращению большей части ацетоацетата в β-гидроксибутират, поэтому основное кетоновое тело в крови — именно β-гидроксибутират. При голодании для многих тканей жирные кислоты и кетоновые тела становятся основными топливными молекулами. Глюкоза используется в первую очередь нервной тканью и эритроцитами.
При высокой концентрации ацетоацетата часть его неферментативно декарбоксилируется, превращаясь в ацетон. Ацетон не утилизируется тканями, но выделяется с выдыхаемым воздухом и мочой. Таким путём организм удаляет избыточное количество кетоновых тел, которые не успевают окисляться, но, являясь водорастворимыми кислотами, вызывают ацидоз.
Рис. 8-32. Активация синтеза кетоновых тел при голодании. Точечные линии — скорость метаболических путей снижена; сплошные линии — скорость метаболических путей повышена. При голодании в результате действия глюкагона активируются липолиз в жировой ткани и (3-окисление в печени. Количество оксалоацетата в митохондриях уменьшается, так как он, восстановившись до малата, выходит в цитозоль, где опять превращается в Оксалоацетат и используется в глюконеогенезе. В результате скорость реакций ЦТК снижается и, соответственно, замедляется окисление ацетил-КоА. Концентрация ацетил-КоА в митохондриях увеличивается, и активируется синтез кетоновых тел. Синтез кетоновых тел увеличивается также при сахарном диабете (см. раздел 11).
Рис. 8-33. Синтез кетоновых тел в митохондриях гепатоцитов. Регуляторный фермент синтеза кетоновых тел (ГМГ-КоА-синтаза) ингибируется свободным КоА. — реакция идёт неферментативно при высокой концентрации кетоновых тел в крови.
Регуляция синтеза кетоновых тел. Регуляторный фермент синтеза кетоновых тел — ГМГ-КоА синтаза.
ГМГ-КоА-синтаза — индуцируемый фермент; его синтез увеличивается при повышении концентрации жирных кислот в крови. Концентрация жирных кислот в крови увеличивается при мобилизации жиров из жировой ткани под действием глюкагона, адреналина, т.е. при голодании или физической работе.
ГМГ-КоА-синтаза ингибируется высокими концентрациями свободного кофермента А.
Когда поступление жирных кислот в клетки печени увеличивается, КоА связывается с ними, концентрация свободного КоА снижается, и фермент становится активным.
Если поступление жирных кислот в клетки печени уменьшается, то, соответственно, увеличивается концентрация свободного КоА, ингибирующего фермент. Следовательно, скорость синтеза кетоновых тел в печени зависит от поступления жирных кислот.
Окисление кетоновых тел в периферических тканях
При длительном голодании кетоновые тела становятся основным источником энергии для скелетных мышц, сердца и почек. Таким образом
глюкоза сохраняется для окисления в мозге и эритроцитах. Уже через 2-3 дня после начала голодания концентрация кетоновых тел в крови достаточна для того, чтобы они проходили в клетки мозга и окислялись, снижая его потребности в глюкозе.
β-Гидроксибутират (рис. 8-34), попадая в клетки, дегидрируется NAD-зависимой дегидрогеназой и превращается в ацетоацетат. Ацетоацетат активируется, взаимодействуя с сук-цинил-КоА — донором КоА:
Ацетоацетат + Сукцинил-КоА → Ацетоацетил- КоА + Сукцинат.
Рис. 8-34. Окисление кетоновых тел в тканях.
Реакцию катализирует сукцинил-КоА-ацето-ацетат-КоА-трансфераза. Этот фермент не синтезируется в печени, поэтому печень не использует кетоновые тела как источники энергии, а производит их «на экспорт». Кетоновые тела — хорошие топливные молекулы; окисление одной молекулы β-гидроксибутирата до СО2 и Н2О обеспечивает синтез 27 молекул АТФ. Эквивалент одной макроэргической связи АТФ (в молекуле сукцинил-КоА) используется на активацию ацетоацетата, поэтому суммарный выход АТФ при окислении одной молекулы β-гидроксибутирата — 26 молекул.
Кетоацидоз. В норме концентрация кетоновых тел в крови составляет 1-3 мг/дл (до 0,2 мМ/л), но при голодании значительно увеличивается. Увеличение концентрации кетоновых тел в крови называют кетонемией, выделение кетоновых тел с мочой — кетонурией. Накопление кетоновых тел в организме приводит к кетоацидозу: уменьшению щелочного резерва (компенсированному ацидозу), а в тяжёлых случаях — к сдвигу рН (некомпенсированному ацидозу), так как кетоновые тела (кроме ацетона) являются водорастворимыми органическими кислотами (рК
3,5), способными к диссоциации:
СН3-СО-СН2-СООН ↔ СН3-СО-СН2-СОО- + Н+.
Ацидоз достигает опасных величин при сахарном диабете, так как концентрация кетоновых тел при этом заболевании может доходить до 400-500 мг/дл. Тяжёлая форма ацидоза — одна из основных причин смерти при сахарном диабете. Накопление протонов в крови нарушает связывание кислорода гемоглобином, влияет на ионизацию функциональных групп белков, нарушая их кон-формацию и функцию.
Гиперкетонемия — это накопление кетоновых тел в крови. К ним относятся ацетоуксусная кислота, р-оксимасляная кислота, ацетон.
Основной причиной гиперкетонемии является увеличение содержания в крови свободных жирных кислот — гиперлипацидемия. Последняя связана с усилением липолиза в жировой ткани, например, при сахарном диабете I типа, лихорадке, во втором периоде голодания.
Избыток свободных жирных кислот поступает в печень, где под действием ферментов р-окисления образуется большое количество ацетил-КоА. Какая-то его часть подвергается дальнейшим превращениям в цикле Кребса (интенсивность этого процесса зависит от энергетических потребностей гепатоцитов), а оставшееся неиспользованное количество идет на образование кетоновых тел, которые поступают в кровь и могут быть утилизированы как ценный энергетический субстрат периферическими клетками.
При некоторых заболеваниях, например, при сахарном диабете I типа, использование кетоновых тел клетками нарушается. Это усугубляет гиперкетонемию.
Гиперкетонемия всегда сопровождается кетонурией (ацетонурией) — появлением кетоновых тел (ацетона) в моче.
Обнаружение кетоновых тел в общем анализе мочи называют — кетонурия. В норме кетоновые тела в моче не обнаруживаются, так как ежедневно выводятся из организма органами выделения, в количестве 25-50 мг в сутки. К таким телам относятся ацетон, уксусная и бета-осмолярная кислоты. В моче здорового человека медики не выявляют кетоновых тел.
Причины появления кетоновых тел
К причинам накапливания в моче кетоновых тел относятся многие причины, некоторые из них несут угрозу нормальной жизнедеятельности организма. Вот одни из причин:
При состояниях, сопровождающихся снижением глюкозы крови, клетки органов и тканей испытывают энергетический голод. Так как окисление жирных кислот процесс «трудоемкий», а нервная ткань вообще неспособна окислять жирные кислоты, то печень облегчает использование этих кислот тканями, заранее окисляя их до уксусной кислоты и переводя последнюю в транспортную форму – кетоновые тела .
К кетоновым телам относят три соединения близкой структуры – ацетоацетат , 3-гидроксибутират и ацетон .
Стимулом для образования кетоновых тел служит поступление большого количества жирных кислот в печень. Как уже указывалось, при состояниях, активирующих липолиз в жировой ткани, не менее 30% образованных жирных кислот задерживаются печенью. К таким состояниям относится голодание , сахарный диабет I типа , длительные физические нагрузки . Так как синтез ТАГ в этих условиях невозможен, то жирные кислоты из цитозоля попадают в митохондрии и окисляются с образованием кетоновых тел.
Кроме отмеченных ситуаций, количество кетоновых тел в крови возрастает при алкогольном отравлении и потреблении жирной пищи. При богатой жирами диете , особенно у детей, жирные кислоты не успевают включиться в состав ТАГ и ЛПОНП и частично переходят в митохондрии, что увеличивает синтез кетоновых тел. При алкогольном отравлении субстратом для синтеза кетонов является ацетил-SКоА, синтезируемый при обезвреживании этанола.
В обычных условиях синтез кетоновых тел также идет, хотя в гораздо меньшем количестве. Для этого используются как жирные кислоты, так и безазотистые остатки кетогенных и смешанных аминокислот.
Для детей раннего возраста характерна неустойчивость жирового обмена, связанная с недостаточностью регуляторного влияния ЦНС и эндокринной системы. Поэтому кетонемия и кетонурия у грудных детей наблюдается чаще, чем у взрослых.
Уровень ацетоацетата быстро нарастает между 12 и 24 часами жизни и сохраняется высоким первую неделю жизни, что свидетельствует об активной мобилизации жира из депо, высокой утилизации свободных жирных кислот в печени и малом использовании кетоновых тел в тканях.
У детей до 7 лет под влиянием различных стимулов (краткое голодание, инфекции, эмоциональное возбуждение) ускоряется синтез кетоновых тел и может легко возникать кетоацидоз, сопровождающийся неукротимой рвотой («ацетонемическая рвота«). Причиной этому служит неустойчивость углеводного обмена и малые запасы гликогена у детей, что усиливает липолиз в адипоцитах, накопление жирных кислот в крови и, следовательно, кетогенез в печени.
Используются кетоновые тела клетками всех тканей, кроме печени и эритроцитов. Особенно активно, даже в норме, они потребляются миокардом и корковым слоем надпочечников.
В тканях реакции утилизации кетоновых тел в целом совпадают с обратным направлением реакций синтеза. В цитозоле клеток 3-гидроксибутират окисляется, образующийся ацетоацетат проникает в митохондрии, активируется за счет сукцинил-SКоА и превращается в ацетил-SКоА, который сгорает в ЦТК.
Главным путем синтеза кетоновых тел, происходящего в основном в печени, считается реакция конденсации между двумя молекулами ацетил-КоА , образовавшегося при β-окислении жирных кислот или при окислительном декарбоксилировании пирувата (пировиноградной кислоты) в процессе обмена глюкозы и ряда аминокислот. Этот путь синтеза кетоновых тел более других зависит от характера питания и в большей степени страдает при патологических нарушениях обмена веществ.
Из печени кетоновые тела поступают в кровь и с нею во все остальные органы и ткани, где они включаются в универсальный энергообразующий цикл — цикл трикарбоновых кислот, в котором окисляются до углекислоты и воды. Кетоновые тела используются также для синтеза Холестерина, высших жирных кислот, фосфолипидов и заменимых аминокислот.
При голодании, однообразном безуглеводистом питании и при недостаточной секреции инсулина использование ацетил-КоА в цикле трикарбоновых кислот подавляется, т.к. все метаболически доступные ресурсы организма превращаются в глюкозу крови. В этих условиях увеличивается синтез кетоновых тел.
При повышении содержания кетоновых тел в крови (нормальное содержание кетоновых тел в плазме крови человека составляет 34,4—430,5 мкмоль/л.) они начинают выводиться с мочой, а также с выдыхаемым воздухом в виде ацетона. Наличие кетоновых тел в моче всегда указывает на развитие патологического состояния. Наиболее значительное повышение концентрации кетоновых в крови (гиперкетонемия) наблюдается при диабетической (кетоацидотической) коме. Интенсивное образование кетоновых тел происходит при приеме с пищей так называемых кетогенных аминокислот (лейцина, тирозина, фенилаланина, изолейцина), некоторых белков и большого количества жиров (при усиленной мобилизации жира из жировых депо). Щелочные соли также проявляют кетогенный эффект, который обусловлен вызываемым ими нарушением функционирования цикла трикарбоновых кислот. Введение с пищей углеводов тормозит образование кетоновых тел. Инсулин стимулирует синтез жирных кислот из ацетил-КоА и активирует использование последнего в цикле трикарбоновых кислот, в результате чего снижается интенсивность синтеза кетоновых тел.
Кетонемию и кетонурию наблюдают при сахарном диабете, углеводном голодании, лихорадочных состояниях, общем голодании и истощении (повышен кетогенез), приеме богатой кетогенными веществами пищи (усилен кетогенез), при приеме значительных количеств щелочных веществ, при состояниях после операций, гликогенозах I, II и VI типа (нарушен кетолиз), гиперинсулинизме, тиреотоксикозе, выраженной глюкозурии, акромегалии, гиперпродукции глюкокортикоидов, инфекционных болезнях (скарлатине, гриппе, туберкулезном менингите и др.) и тяжелых интоксикациях (например, при отравлении свинцом) и др. Следствием кетонемии являются метаболический Ацидоз, или кетоацидоз, и ацетоновое отравление (ацетон растворяет структурные липиды клеток), при которых нарушается транспорт глюкозы через биологические мембраны и резко угнетается деятельность ц.н.с.
• сахарный диабет (вариант метаболического ацидоза, связанный с нарушением углеводного обмена, возникшего вследствие дефицита инсулина: высокой концентрацией глюкозы и кетоновых тел в крови (значительно превышающей физиологические значения), образованных в результате нарушения обмена жирных кислот (липолиза) и дезаминирования аминокислот. Если нарушения углеводного обмена не купируются своевременно — развивается диабетическая кетоацидотическая кома.)
26.ЗАДАЧА. В крови обнаружено значительное повышение индикаторных ферментов АЛТ, АСТ, ФМФА (фруктозомонофосфатальдолазы), повышение содержания билирубина; щелочная фосфатаза и g-глутамилтранспептидаза в пределах нормы, содержание белка в норме. Какой тип синдрома имеет место? Какие функции печени нарушены?
Такие синдромы характены для цитолиза. Сущность процесса цитолиза заключается в разрушении клеточной мембраны гепатоцита. Цитолиз гепатоцитов начинается с локальных повреждений поверхности клетки, ведущих к нерегулируемой проницаемости клеточной мембраны как для межклеточной среды, так и для молекулярных компонентов самой клетки. В результате осмотического электролитного дисбаланса происходит задержка жидкости в гепатоцитах и их набухание. Изменяется уровень физиологических концентраций реагирующих веществ, нарушается регуляция активности ферментных систем в цитоплазме. Таким образом, частичные нарушения целостности клеточных мембран приводят к расстройству внутриклеточного метаболизма, приостановке развития и роста гепатоцитов. Накопление повреждений гепатоцитов приводит к нарастанию функциональной блокировки печени, как целого органа, при ее кажущейся целостности. Дальнейшее распространение процесса дезинтеграции мембран клеток на внутриклеточные органеллы, разрушение их стенок окончательно расстраивает внутриклеточные обменные процессы и освобождает активные гидролитические ферменты лизосом, разрушающие основные компоненты клетки: белки, нуклеиновые кислоты и др.
27.ЗАДАЧА. У больного желтухой в крови обнаружено незначительное повышение АЛТ, АСТ, значительное повышение активности щелочной фосфатазы и g-глутамил-транспептидазы, повышено содержание холестерина и прямого билирубина, содержание общего белка в пределах нормы. Какой тип синдрома имеет место, причины его возникновения?
Синдром холестаза обусловлен нарушением желчевыделительной функции печеночных клеток с нарушением образования желчной мицеллы и поражением мельчайших желчных протоков. Морфологической основой синдрома холестаза является застой желчи в желчевыводящих путях с последующей вторичной дистрофией гепатоцитов.
30. . ЗАДАЧА. В план обследования больного с фурункулезом включается глюкозо-толерантный тест. Опишите методику проведения этого теста. Интерпретация полученных результатов.
Тест на устойчивость к нагрузке глюкозой проводится с определением уровня глюкозы по трем точкам: до нагрузки, через 1 час и через 2 часа после нагрузки. Провидится, если не ясен диагноз. Выполнение: пациент поддерживает обычное питание в течении 3 дней. Углеводов должно быть примерно 250 гр. Тест выполняют утром, натощак. Измеряют уровень глю. Затем больному дают примерно 75 гр глю, растворенной в чае с лимоном. Затем берут пробы через 60 мин и 120 мин. Результаты при фурункулезе : натощак-уровень глю в норме, через час- превышает норму(глю появляется в моче), через 2 ч глю соответствует норме. При анализе результатов только натощак и через 2 часа нарушений не наблюдается. Жалобы не наблюдаются.
Задача №29 В основе механизма развития Диабет стероидный лежат воздействия глюкокортикоидов на белковый и углеводный обмен. Они усиливают распад белков и тормозят их синтез. При увеличенном выделении аминокислот из тканей и поступлении их в печень значительно ускоряется процесс переаминирования, далее дезаминирования аминокислот, которые используются для процессов глюконеогенеза. В печени увеличивается отложение гликогена. Повышение выделения азота с мочой под влиянием глюкокортикоидов указывает, что источником описанных нарушений является распад белков. Воздействие глюкокортикоидов на углеводный обмен проявляется усилением активности глюкозо-6-фосфатазы и торможением активности глюкокиназы печени. Антиинсулиновый эффект стероидов на периферии проявляется уменьшением утилизации глюкозы тканями. Глюкокортикоиды усиливают липогенез. Антикетогенные свойства глюкокортикоидов связаны с угнетением окисления пировиноградной к-ты, отсюда увеличение в крови молочной кислоты
Механизм глюкозурии.
В начальном (проксимальном) отделе канальца глюкоза реабсорбируется из первичной мочи, проходя через эпителий, выстилающий канальцы, назад в кровоток. Проблема в том, что проксимальные канальцы способны реабсорбировать лишь ограниченное количество глюкозы. Дело в том, что для реабсорбции глюкозы необходимо связывание каждой ее молекулы с молекулой переносчика, поэтому транспорт глюкозы является насыщаемым. Когда гликемия превышает некоторый критический уровень (обычно 8,9-10,0 ммоль/л или 160-180 мг/дл), проксимальные канальцы оказываются «перегруженными» — а весь излишек глюкозы попадает во вторичную (выделяемую в мочевой пузырь) мочу.
Эта критическая точка получила условное название «почечный порог». Он индивидуален для каждого человека, но, как правило, укладывается в вышеуказанный диапазон концентрации глюкозы крови.
Синдром: гепатоцеллюлярная недостаточность
Играют важную роль в оценке острого вирусного гепатита, хронического активного гепатита (ХАГ) и цирроза печени:
Тимоловая (тимолвероналовая) проба. Норма: О—7 ед. по Маклагану, 3—30 ед. по Венсану.
Сулемовая проба. Норма: 1,9 ед. и выше.
Индикаторы холестатического синдрома тонко регистрируют нарушения секреции и циркуляции желчи, однако не могут дифференцировать внутрипеченочные и подпеченочные формы холестаза. Выделение этих форм ХС производится только с помощью ретроградной хо-лангиографии и УЗ И желчных путей.
Щелочная фосфатаза -Норма: 2—5 ед.
Гамма-глутамилтрансфераза (ГГТФ) Норма: для мужчин 15—106 усл. ед.,для женщин 10—66 усл. ед.,
Билирубин Норма: общий — ниже 1,2 мг/100мл, или 20,5 ммоль/л; прямой (конъюгированный) — 0,86—4,3 мкмоль/л, не более 25% от общего; непрямой (неконъю-гированный) — 1,7—17,1 мкмоль/л, 75% и более от общего.
Индикаторы цитолитического синдрома представлены в основном рядом ферментов сыворотки крови.
Аспартатаминотрансфераза(АсАТ). Норма: 7—40 усл. Ед
Аланинаминотрансфераза (АлАТ). Норма: 7—40 усл. ед.
Гамма-глутамилтрансфераза ГГТФ, Норма: для мужчин 15—106 усл. ед., для женщин 10—66 усл. ед.,
Глутаматдегидрогеназа ГлДГ. Норма: 0—0,9 усл. ед.
Лактатдегидрогеназа ЛДГ. Норма: 100—340 усл. ед., 0,8—4 мкмоль/мл,
При заболеваниях печени происходит нарушение синтеза эфиров холестерина и, естественно, уменьшение их количества в крови. Общее количество холестерина уменьшается лишь при тяжелых поражениях печени .Повышение количества холестерина в крови характерно для заболеваний, сопровождающихся нарушением экскреции желчи (механическая желтуха, билиарный цирроз).
Общее количество холестерина в сыворотке крови составляет 150—200 мг%, из этого количества 2/3 приходится на его эфиры.
Принцип метода определения активности дегидрогеназ. Примеры специфических ингибиторов дегидрогеназ и механизмы их действий.
Дегидрогеназы относятся к числу так называемых «тиоловых» ферментов, каталитическое действие которых обусловлено сульфгидрильными (тиоловыми) группами, входящими в состав активного центра молекул фермента или ответственными за поддержание каталитически активной конформации . Поэтому ингибиторами дегидрогеназ являются вещ-ва, блокирующие SH-группы,к ним относятся окисляющие, меркаптидобразующие алкилирующие вещ-ва, такие как перекись водорода(окислитель),хлорид кадмия и др. тяжелые металлы(Hg,Pb,Ag,Cu-меркаптидобразующий агент).Ингибирование протекает по уравнению реакций:
2) белок-S-Cd-Cl+ Белок-SH——> белок-S-Cd-S-белок+ HCl
3) Белок-SH+ICH2COOH—-> Белок-SH-CH2COOH+HI
Метод определения активности дегидрогеназ может быть использован в гигиенических исследованиях при производственных и пищевых отравлениях окислителями ,солями тяжелых металлов ,алкилирующими агентами. Активность дегидрогеназ определяется при экспериментальных и клинических исследованиях, направленных соответственно либо на изучение биохимических мех-ов действия на организм химических и физических факторов среды(ионизирующая и ультрафиолетовая радиация, шум, вибрация),либо на выявление признаков интоксикаций и др. заболеваний.
Какие методы биохимических исследований и почему могут быть использованы для контроля за состоянием здоровья лиц, контактирующих с окислителями, тяжелыми металлами, галогенопроизводными?
Эти вещества являются ингибиторами дегидрогеназ. Метод определения активности дегидрогеназ может быть использован в гигиенических исследованиях при производственных и пищевых отравлениях окислителями ,солями тяжелых металлов ,алкилирующими агентами. Активность дегидрогеназ определяется при экспериментальных и клинических исследованиях, направленных соответственно либо на изучение биохимических мех-ов действия на организм химических и физических факторов среды(ионизирующая и ультрафиолетовая радиация, шум, вибрация),либо на выявление признаков интоксикаций и др. заболеваний.
Какие биохимические изменения характерны для диабета?
Задача. Биохимические тесты используемые для дифференциальной диагностики инсулинзависимого и инсулиннезависимого сахарного диабета.
Инсулинзависимый диабет (сахарный диабет 1 типа) развивается в основном у детей и молодых людей;
Инсулиннезависимый диабет (сахарный диабет 2 типа) обычно развивается у людей старше 40 лет, имеющих избыточный вес. Это наиболее распространенный тип болезни (встречается в 80-85% случаев);
При сахарном диабете 1 типаимеется абсолютный дефицит инсулина, обусловленный нарушением работы поджелудочной железы.
При сахарном диабете 2 типаотмечаетсяотносительный дефицит инсулина. Клетки поджелудочной железы при этом вырабатывают достаточно инсулина (иногда даже повышенное количество). Однако на поверхности клеток блокировано или уменьшено количество структур, которые обеспечивают его контакт с клеткой и помогают глюкозе из крови поступать внутрь клетки. Дефицит глюкозы в клетках является сигналом для еще большей выработки инсулина, но это не дает эффекта, и со временем продукция инсулина значительно снижается.
Обязательным при диагностике и лечении сахарного диабета является определение суточной глюкозурии – количество сахара, выделяемого в суточной порции мочи. При установленном диабете глюкозурию определяют с целью отслеживания эффективности назначенного лечения, критерием которого является снижение суточной глюкозурии. Используют специальные экспресс тест-полоски.
Исследование уровня глюкозы в крови проводится натощак и может совмещаться с анализом на сахар суточной мочи. Если по результату анализа крови количество сахара выше 120мг%, сахарный диабет считается установленным. Если полученные результаты укладываются в норму, назначается анализ на гликированный гемоглобин. С его помощью можно определить количество гемоглобина с присоединенными к нему молекулами глюкозы – гликированного гемоглобина. Этот анализ позволяет установить средний уровень глюкозы в крови пациента за 2-3 месяца до его проведения и необходим для определения скрытой формы сахарного диабета.
В том случае, когда уровень гликированного гемоглобина высокий, проводится тест на толерантность к глюкозе, или анализ на уровень глюкозы в крови после нагрузки – употребления определенного количества сахара. В норме у здорового человека после употребления сахара повышается количество глюкозы, а затем достаточно быстро возвращается к исходным данным. При сахарном диабете, даже при скрытой его форме, после сахарной нагрузки уровень глюкозы в крови резко возрастает и через два часа продолжает оставаться высоким. Поэтому анализ крови на уровень глюкозы с нагрузкой очень важный момент в диагностике сахарного диабета и преддиабетических состояний.
Такое исследование проводится утром, после ночного воздержания от пищи не менее 12 часов. В течение трех дней до анализа пациент не должен придерживаться какой-либо диеты, а употреблять привычные продукты и испытывать обычные нагрузки. В день теста запрещается курить, испытывать стресс, употреблять некоторые лекарственные препараты. Сначала определяется исходный уровень глюкозы в крови, затем пациенту предлагается 75г глюкозы, растворенной в 300мл воды, и через 2 часа после ее приема проводят повторный анализ.
Нормальные показатели глюкозы в крови:
| Возраст | Уровень глюкозы, ммоль/л |
| 70 лет | 4,61 — 6,10 |
ЗАДАЧА 14. Каковы причины нарушения переваривания белков в желудочно-кишечном тракте.
Задача. Какие нарушения в организме могут возникнуть при нарушении выделения желчных кислот.
Основные причины недостаточного расщепления белков заключаются в количественном уменьшении секреции соляной кислоты и ферментов, снижении активности протеолитических ферментов (пепсина, трипсина, химотрипсина) и связанном с этим недостаточным образованием аминокислот, уменьшении времени их воздействия (ускорение перистальтики)- Так, при ослаблении секреции соляной кислоты снижается кислотность желудочного сока, что ведет к уменьшению набухания пищевых белков в желудке и ослаблению превращения пепсиногена в его активную форму — пепсин. В этих условиях часть белковых структур переходит из желудка в двенадцатиперстную кишку в неизмененном состоянии, что затрудняет действие трипсина, химотрипсина и других протеолитических ферментов кишечника.
Недостаточное образование свободных аминокислот из пищевых белков может происходить в случае ограничения поступления в кишечник сока поджелудочной железы (при панкреатите, сдавлении, закупорке протока). Недостаточность функции поджелудочной железы ведет к дефициту трипсина, химотрипсина, карбоангидразы А, Б и других протеаз, воздействующих на длинные полипептидные цепи или расщепляющих короткие олигопептиды, что снижает интенсивность полостного или пристеночного пищеварения.
Недостаточное действие пищеварительных ферментов на белки может возникнуть вследствие ускоренного прохождения пищевых масс по кишечнику при усилении его перистальтики (при энтероколитах) либо уменьшении площади всасывания (при оперативном удалении значительных участков тонкого кишечника). Это ведет к резкому сокращению времени контакта содержимого химуса с апикальной поверхностью энтероцитов, незавершенности процессов энзиматического распада, а также активного и пассивного всасывания.
Нарушения биосинтеза желчных кислот наиболее выражены при циррозе печени, когда вследствие понижения активности 12α-гидроксилазы наблюдается уменьшенное образование холевой кислоты. Нарушение синтеза холевой кислоты ведет за собой нарушение ее превращения в дезоксихолевую кислоту. И хотя при циррозе печени биосинтез хенодезоксихолевой кислоты протекает нормально, общий запас желчных кислот за счет холевой и дезоксихолевой кислот уменьшается примерно на 50%. Вследствие снижения объема их циркуляции уменьшается концентрация желчных кислот в тонком кишечнике при приеме пищи. Таким образом, резорбция жирорастворимых витаминов и жиров нарушается, что провоцирует гипо- или авитаминоз витаминов А, D, K и, в частности, явления остеомаляции, нарушений свертывания крови и стеатореи.
Задача.Цианиды-являются ингибиторами цитохромоксидазы аа3 в дыхательной цепи. Тип ингибирования-необратимый. Восстановить дыхательную цепь невозможно, блокируется образование АТФ, возникает состоянии гипоксии.
Механизм развития метгемоглобинемий: Метгемоглобин образуется в результате окисления железа в геме гемоглобина, то есть метгемоглобинообразование идет под действием любых веществ с дефицитов электронов.

Если содержание метгемоглобина превышает 3% -это называется метгемоглобинемия.
Бывают врожденные и приобретенные метг-мии.
Врожденная имеет место при энзимопатиях и гемоглобинозах Энзимопатии-генетические дефекты синтеза ключевых ферментов, которые участвуют в восстановлении MеtHb или ключевых ферментов ПФЦ, генерирующий НАДФН. Гемоглобинозы делят на гемоглобинопатии –генетические нарушения строения белкой части Hb, возникающие в результате точечных мутаций ,и талассемии-нарушение синтеза какой-либо цепи Hb.
Приобретенные бывают эндогенные и экзогенные. Эндогенная связанная с недостаточностью АОС,экзогенная возникает в случае избыточного поступления окислителей. Метгемоглобин теряет способность транспорта кислорода, и возникает гипоксия
Если вовремя не оказать помощь возникает смерть от недостаточности кислорода, для помощи пострадавшему необходимо повысить парциальное давление O2(вдыхание чистого O2)
21.ЗАДАЧА. Мочевина, происхождение, причины изменения содержания в крови и моче.
Мочевина-конечный продукт обмена белков. Синтезируется в гепатоцитах в орнит.цикле из аммиака при участии СО2.данный процесс треб.затраты энергии, поэтому важным условием синтеза мочевины явл.достат.снабжение печени кислородом. Далее мочевина поступает в кровь и выводится с мочой ч\з почки (12-36г\сут.).
· Повышение содержания мочевины в крови связано с поражением почек (нарушена почечная фильтрация), при этом ее содерж.в моче понижено.
· При повреждении гепатоцитов будет понижение синтеза мочевины, и следовательно ее содержание в крови и моче.
· Рост мочевины и в крови и в моче наблюд.при усилении распада тканевых белков (сах.диабет, онкология).
Норма мочевины в крови: 2,8-8,3 ммоль\л, в моче: 330-580 ммоль\сут. Повышение мочевины в моче – азотурия.
Повыш. Конц.может быть и физиологическим например при диете с большим потреблением белков или при беременности.
Более редкие причины: атрофия мышц, отравление фосфором, гепатиты, недостаток вит.Е и В1, дефицит селена, нарушение гормонального баланса.
*У больного с желтухой в крови обнаружено: активность АСТ и АЛТ
на нижней границе нормы; активность сывороточной холинэстеразы снижена значительно, снижено содержание общего белка, особенно альбуминов, фибриногена, холестерина, повышено содержание общего билирубина. Какой тип синдрома поражения печени имеет место? Какие функции печени нарушены?
Коэффициент активности АСТ\АЛТ=1, при циррозе он выше, при гепатите ниже.
Общий билирубин: 8,5-20,5 мкмоль\л
Все показатели свидетельствуют о наличии воспалит.процесса в печени – гепатита! Повреждена паренхима печени,что вызывает нарушение пищеварительной функции организма, поэтому человек болеющий гепатитом страдает неспособностью кишечника полностью впитывать питательные вещества. Также при гепатите нарушается барьерная функция печени, и в кровь вместе с вирусом гепатита поступают ядовитые вещества. Процесс «самоотравления» при гепатите происходит всегда. При гепатите печень занята борьбой с вирусом гепатита, поэтому другие вирусы и бактерии способны оседать в организме. Поражая печень, гепатит не дает ей нейтрализовать яды разлагающихся белков, непереваренных также из-за гепатита. Таким образом гепатит вызывает общее отравление организма. При поражении печени гепатитом всегда возникает дискинезия желчевыводящих путей, таким образом гепатит приводит к образованию билирубиновых камней. Пораженная гепатитом печень не способна разлагать лишний холестерин, поэтому у больных гепатитом образуются холестериновые камни. У больных гепатитом эфиры холестерина откладываются во всём организме.
25.ЗАДАЧА. Как изменится содержание креатина и креатинина в крови и моче при: а) миопатии, б) заболевании почек, в) усиленной мышечной работе.
Норма креатинина в моче: 4,4-17,7 ммоль\сут, в крови: 0,044-0,1 ммоль\л
Норма креатина в моче : до 380 мкмоль\сут, в крови:
а) при миопатии креатинин в моче понижен,в крови повышен. Креатин в моче повышен
б) при заболеваниях почек креатинин в моче понижен,в крови повышен. Креатин в моче может быь повышен или понижен,в зависимости от диагноза.
В) при интенсивной мышечной работе креатинин в моче и в крови повышен.
1. Задача. О поражениях каких органов может идти речь при наличии следующего спектра крови:
1) АлАТ > АсАТ > ГлДГ > амилаза.
2) КФК > АсАТ > ЛДГ > амилаза > ГлДГ.
3) амилаза > липаза > АлАТ > АсАТ > КФК
В статье представлены современные данные о физиологии энергетического обмена и роли в нем кетоновых тел. Рассмотрены основные причины избыточного образования кетонов, методы диагностики, подходы к лечению.
The article presents modern data on the physiology of energy metabolism and the role of ketone bodies in it. The main causes of excessive ketone formation, diagnostic methods, approaches to treatment are considered.
Часть 2. Начало статьи читайте в № 6, 2017 г.
Голодание — это состояние организма, связанное с частичным или полным нарушением поступления пищи. В состоянии голодания резко снижаются источники энергии организма для важнейших структур организма. В условиях дефицита питательных веществ в организме образование энергии происходит за счет интенсификации глюкогенеза и синтеза кетоновых тел. Содержание глюкозы в крови уменьшается до нижних пределов нормы (3,5 ммоль/л) и на этом уровне поддерживается и в последующие периоды голодания. В печени при голодании глюкоза не в состоянии обеспечить должного количества оксалоацетата, поскольку ее просто нет в клетке. Поэтому при голодании жирные кислоты не «сгорают» в ЦТК, а превращаются в кетоновые тела.
Снижение запасов гликогена в печени сопровождается усиленным поступлением в нее свободных жирных кислот из адипоцитов. Концентрация жирных кислот в крови увеличивается в 3–4 раза по сравнению с постабсорбтивным состоянием. Уровень кетоновых тел в крови через неделю голодания повышается в 10–15 раз. В то же время дефицит углеводов тормозит окисление кетоновых тел, замедляя ресинтез их в высшие жирные кислоты [13].
Энергетические потребности мышц и большинства других органов удовлетворяются за счет жирных кислот и кетоновых тел. При низком уровне инсулина глюкоза в мышечные клетки не проникает, потребителями глюкозы являются инсулинонезависимые клетки и прежде всего клетки мозга, но и в этой ткани биоэнергетика частично обеспечивается кетоновыми телами. При такой концентрации ацетоуксусная кислота активно декарбоксилируется с образованием ацетона, который выводится с выдыхаемым воздухом и через кожу. Уже на 3–4 день изо рта и от кожи голодающего исходит запах ацетона.
Организм включает альтернативные способы выработки энергии — это глюконеогенез и синтез кетокислот, которые потребляются центральной нервной системой. При голодании повышается выброс глюкагона, который активирует липолиз в адипоцитах и окисление в печени. Количество оксалоацетата в митохондриях уменьшается, так как он, восстановившись до малата, выходит в цитозоль клетки, где опять превращается в оксалоацетат и используется в глюконеогенезе.
Глюконеогенез продолжается за счет распада тканевых белков. Аминокислоты образуются в результате распада мышечных белков и включаются в глюконеогенез при длительном голодании. Пируват образуется в печени из лактата и аланина. Аланин и глутамин являются наиболее важными глюкогенными аминокислотами при голодании. Пируват и метаболиты ЦТК способны образовывать оксалоацетат и включаться в глюконеогенез.
При голодании подавляется использование ацетил-КоА в ЦТК, и он используется исключительно для синтеза оксиметилглутарил-КоА, что приводит к увеличению образования кетоновых тел. В этих условиях кетоновые тела являются альтернативным (глюкозе) энергетическим материалом для мозга и других тканей. 75% потребности мозга в энергии удовлетворяется за счет ацетил-КоА [4].
Если голодание продолжается дни, недели — включаются другие гомеостатические механизмы, которые обеспечивают сохранение белковой структуры организма, замедляя глюконеогенез и переключая мозг на утилизацию кетоновых молекул. Сигналом для использования кетонов служит повышение их концентрации в артериальной крови. При длительном голодании наблюдаются крайне низкие концентрации инсулина в крови. В этом случае интенсивный кетогенез представляет собой компенсаторно приспособительную реакцию.
Интенсивность обмена веществ в целом снижена: через неделю голодания потребление кислорода уменьшается примерно на 40%, происходят торможение окислительных процессов в митохондриях и угнетение окислительного фосфорилирования с образованием АТФ, т. е. развивается гипоэнергетическое состояние.
Накапливаясь в крови, кетоновые тела подавляют секрецию и активность глюкокортикоидов, тем самым препятствуя разрушению структурных белков организма и угнетая секрецию глюкагона [2]. Если в это время голодающему вводить аланин или другие гликогенные аминокислоты, уровень глюкозы в крови повышается, а концентрация кетоновых тел снижается.
При голодании кетоз опасности не представляет, так как не достигает степени кетоацидоза. Последний развивается при сопутствующих факторах — дегидратации, алкогольной интоксикации и других состояниях.
Гиперпродукция кетокислот и кетоацидоз после чрезмерного употребления спиртного частое наблюдаемое состояние. Катаболизм этилового спирта осуществляется главным образом в митохондриях печени. Здесь окисляется от 75% до 98% введенного в организм этанола. Окисление алкоголя — сложный биохимический процесс. Основную роль в метаболизме этанола играет никотинамидадениндинуклеотид (NAD). Этот фермент превращает этанол в токсический метаболит — ацетальдегид и восстановленный NADH, а последний соответствует синтезу ацетоацетата и β-оксибутирата.
Алкогольдегидрогеназа катализирует обратимую реакцию, направление которой зависит от концентрации ацетальдегида и соотношения NADH/NAD + в клетке. Повышение концентрации ацетальдегида в клетке вызывает индукцию фермента альдегидоксидазы. В ходе реакции образуются уксусная кислота.
Полученная в ходе реакции уксусная кислота активируется под действием фермента ацетил-КоА-синтетазы. Реакция протекает с использованием кофермента А и молекулы АТФ. Образовавшийся ацетил-КоА, в зависимости от соотношения АТФ/АДФ и концентрации оксалоацетата в митохондриях гепатоцитов, может «сгорать» в ЦТК или использоваться на синтез жирных кислот или кетоновых тел.
На начальных стадиях алкоголизма ацетил-КоА в ЦТК — основной источник энергии для клетки. Избыток ацетил-КоА в составе цитрата выходит из митохондрий, и в цитоплазме начинается синтез жирных кислот.
В период острой алкогольной интоксикации, несмотря на наличие большого количества ацетил-КоА, недостаток оксалоацетата снижает скорость образования цитрата. В этих условиях избыток ацетил-КоА идет на синтез кетоновых тел. Увеличение концентрации NADH по сравнению с NAD + замедляет реакцию окисления лактата, увеличивается соотношение лактат/пируват. В крови возрастает концентрация лактата, это приводит к гиперлактацидемии и лактоацидозу. Повышение в крови содержания лактата, ацетоуксусной кислоты и β-гидроксибутирата служит причиной метаболического ацидоза при алкогольной интоксикации [14].
Способствует усиленному кетогенезу при алкогольной интоксикации гипогликемические состояния, связанные с рвотой и голоданием. Известно также, что у таких пациентов уровень инсулина в крови снижен, тогда как содержание кортизола, гормона роста, глюкагона и адреналина повышено. Этанол тормозит глюконеогенез. Дегидратация в этих случаях способствует кетогенезу.
На уровень глюкозы в крови влияет широкий спектр гормонов, при этом только инсулин вызывает гипогликемический эффект. Контринсулярным действием с повышением уровня глюкозы крови обладают все гормоны: глюкагон, адреналин, глюкокортикоиды, адренокортикотропный (АКТГ), соматотропный (СТГ), тиреотропный (ТТГ), тиреоидные.
Эффекты инсулина и контринсулярных гормонов в норме регулируют стабильный уровень глюкозы в крови. При низкой концентрации инсулина усиливаются гипергликемические эффекты других гормонов, таких как глюкагон, адреналин, глюкокортикоиды и гормон роста. Это происходит даже в том случае, если концентрация этих гормонов в крови не увеличивается.
Патогенез кетоза при избытке тироксина, глюкокортикоидов, соматотропина или/и других гормонов, в сущности, аналогичен уже рассмотренным механизмам гиперпродукции кетокислот вследствие избытка контринсулярных гормонов [6]. Известно, что в период усиленного роста, а также при гипертиреозе наступает значительное похудание.
При стрессе активируется симпатическая нервная система и выброс контринсулярных гормонов, происходит истощение углеводных резервов организма, нарушается способность печени синтезировать и откладывать гликоген. Происходит избыточное поступление в печень неэтерифицированных жирных кислот. В результате повышенной продукции глюкокортикоидов идет распад белков и усиленное образование кетоновых тел из кетогенных аминокислот.
Гиперкортицизм
Ацетонемический синдром может быть первым клиническим проявлением гиперкортицизма, когда характерные признаки заболевания еще не сформировались.
Глюкокортикоиды способствуют усилению мобилизации нейтральных жиров из жировой ткани и тормозят липогенез. Но это действие в организме может перекрываться другими эффектами данных гормонов: способностью вызывать гипергликемию и стимулировать секрецию инсулина, накопление гликогена в печени, что приводит к торможению мобилизации жира и его отложению в жировой ткани; способностью в больших дозах задерживать жиромобилизующее и стимулирующее окисление жиров соматотропином.
Этим можно объяснить накопление жира в жировых депо при гиперкортицизме (болезни и синдроме Иценко–Кушинга). Кроме того, при этом состоянии увеличено образование дигидрокортизона, который стимулирует пентозный цикл и превращение углеводов в жиры. Кортикотропин, стимулируя секрецию глюкокортикоидов, может влиять на жировой обмен в том же направлении, но, помимо этого, обладает еще и экстраадреналовым жиромобилизующим действием [6].
Тиреотоксикоз
Избыток тиреоидных гормонов в крови может быть следствием заболеваний, проявляющихся гиперфункцией щитовидной железы. Тяжелым осложнением основного заболевания, сопровождающегося гиперфункцией щитовидной железы, является тиреотоксический криз, который представляет собой резкое обострение всех симптомов тиреотоксикоза. Чрезмерное поступление в кровь тироидных гормонов вызывает тяжелое токсическое поражение сердечно-сосудистой системы, печени, нервной системы и надпочечников. В клинической картине характерны резкое возбуждение (вплоть до психоза с бредом и галлюцинациями), которое затем сменяется адинамией, сонливостью, мышечной слабостью, апатией. Усиливаются диспепсические расстройства: жажда, тошнота, рвота, жидкий стул. Возможно увеличение печени. На этом фоне резко усиливаются процессы кетогенеза, что может спровоцировать симптомы ацетонемии.
Тироксин обладает жиромобилизующим эффектом. При гипертиреозе усилен обмен углеводов. Увеличена утилизация глюкозы тканями. Активируется фосфорилаза печени и мышц, следствием чего является усиление гликогенолиза и обеднение этих тканей гликогеном. Увеличивается активность гексокиназы и всасывание глюкозы в кишечнике, что может сопровождаться алиментарной гипергликемией. Активируется инсулиназа печени, что вместе с гипергликемией вызывает напряженное функционирование инсулярного аппарата и в случае его функциональной неполноценности может привести к развитию сахарного диабета. Усиление пентозного пути обмена углеводов способствует образованию НАДФ-Н2. В надпочечниках это вызывает повышение стероидогенеза и большее образование кортикостероидов [4].
Дефицит гормонов
Гипогликемия всегда встречается при пангипопитуитаризме — заболевании, характеризующемся снижением и выпадением функции передней доли гипофиза (секреции адренокортикотропина, пролактина, соматотропина, фоллитропина, лютропина, тиреотропина). В результате резко снижается функция периферических эндокринных желез. Однако гипогликемия встречается и при первичном поражении эндокринных органов (врожденная дисфункция коры надпочечников, болезнь Аддисона, гипотиреоз, гипофункция мозгового слоя надпочечников, дефиците глюкагона). При дефиците контринсулярных гормонов снижается скорость глюконеогенеза в печени (влияние на синтез ключевых ферментов), повышается утилизация глюкозы на периферии, снижается образование аминокислот в мышцах — субстрата для глюконеогенеза.
Дефицит глюкокортикоидов
Первичная надпочечниковая недостаточность является следствием уменьшения секреции гормонов коры надпочечников. Под этим термином подразумевают различные по этиологии и патогенезу варианты гипокортицизма. Симптомы надпочечниковой недостаточности развиваются только после разрушения 90% объема ткани надпочечников.
Причины гипогликемии при надпочечниковой недостаточности схожи с причинами гипогликемии при гипопитуитаризме. Отличием является уровень возникновения блока — при гипопитуитаризме снижается секреция кортизола из-за дефицита АКТГ, а при надпочечниковой недостаточности из-за разрушения ткани самих надпочечников.
Гипогликемические состояния у больных с хронической надпочечниковой недостаточностью могут возникать как натощак, так и через 2–3 часа после приема пищи, богатой углеводами. Приступы сопровождаются слабостью, чувством голода, потливостью. Гипогликемия развивается в результате снижения секреции кортизола, уменьшения глюконеогенеза, запасов гликогена в печени.
Дефицит катехоламинов
Данное состояние может возникать при надпочечниковой недостаточности с поражением мозгового слоя надпочечников. Катехоламины, попадая в кровь, регулируют высвобождение и метаболизм инсулина, снижая его, а также увеличивают высвобождение глюкагона. При снижении секреции катехоламинов наблюдаются гипогликемические состояния, вызванные избыточной продукцией инсулина и пониженной активностью гликогенолиза.
Дефицит глюкагона
Глюкагон — гормон, являющийся физиологическим антагонистом инсулина. Он участвует в регуляции углеводного обмена, влияет на жировой обмен, активируя ферменты, расщепляющие жиры. Основное количество глюкагона синтезируется альфа-клетками островков поджелудочной железы. Однако установлено, что специальные клетки слизистой оболочки двенадцатиперстной кишки и слизистой оболочки желудка также синтезируют глюкагон. При поступлении в кровоток глюкагон вызывает повышение в крови концентрации глюкозы, вплоть до развития гипергликемии. В норме глюкагон предотвращает чрезмерное снижение концентрации глюкозы. Благодаря существованию глюкагона, препятствующего гипогликемическому действию инсулина, достигается тонкая регуляция обмена глюкозы в организме.
При дефиците вышеперечисленных гормонов содержание инсулина снижено, а экскреция кетоновых тел с мочой повышена [4].
Печень участвует в поддержании нормального уровня глюкозы в сыворотке крови путем гликогеногенеза, гликогенолиза и глюконеогенеза. В основе нарушений обмена углеводов при болезнях печени лежат повреждения митохондрий, которые ведут к снижению окислительного фосфорилирования. Вторично страдают функции печени. При тяжелом остром гепатите, как правило, отмечается гипогликемия, а при циррозах печени это наступает в конечной стадии — при печеночной недостаточности [15]. Гипогликемия объясняется снижением способности печени (из-за обширного поражения ее паренхимы) синтезировать гликоген и уменьшением выработки инсулиназы (фермента, разрушающего инсулин).
Дефицит углеводов приводит также к усилению анаэробного гликолиза, вследствие чего в клетках накапливаются кислые метаболиты, вызывающие снижение рН. При циррозе печени может повышаться и уровень лактата в сыворотке крови в связи со сниженной способностью печени утилизировать его для глюконеогенеза.
При заболеваниях печени увеличивается роль жиров в качестве источника энергии. В печени происходят синтез жирных кислот и их расщепление до ацетил-КоА, а также образование кетоновых тел, насыщение ненасыщенных жирных кислот и их включение в ресинтез нейтральных жиров и фосфолипидов. Катаболизм жирных кислот осуществляется путем β-окисления, основной реакцией которого является активирование жирной кислоты с участием кофермента ацетил-КоА и АТФ. Освобождающийся ацетил-КоА подвергается полному окислению в митохондриях, в результате чего клетки обеспечиваются энергией.
При ряде заболеваний печени снижается и синтез липопротеидов, что ведет к накоплению триацилглицеридов с последующей инфильтрацией и жировой дистрофией печени. Причинами возникновения этого состояния, в частности, является недостаток в пище липотропных веществ (холина — составной части лецитина, метионина). Увеличивается образование кетоновых тел [4].
Итак, клиническая картина вторичного ацетонемического синдрома включает в себя непосредственно явления кетоза, признаки основного заболевания, на фоне которого развился кетоз, а также проявления того состояния, которое запустило патологический процесс (стресс, чрезмерная физическая нагрузка, инфекция и т. д.).
В практике приходится сталкиваться с идиопатической ацетонемической рвотой, которая протекает с кетоацидозом (ацетонемическая рвота, недиабетический кетоацидоз). В англоязычной литературе она входит в синдром идиопатической циклической рвоты [16, 17].
Патогенез ацетонемической рвоты полностью не выяснен. Предполагается, что у детей после перенесенных инфекционных заболеваний, травм черепа, органических заболеваний центральной нервной системы в течение длительного времени в гипоталамо-диэнцефальной области остается доминантный очаг застойного возбуждения, индуцирующий нарушения жирового обмена (усиление кетогенеза, нарушение нормального использования кетоновых тел в связи с истощением углеводных запасов в организме). В патогенезе ацетонемической рвоты могут иметь значение аномалии конституции, относительная несостоятельность энзимных систем печени, нарушения эндокринной регуляции метаболизма.
Перспективными являются представления о синдроме циклической рвоты как о митохондриальной патологии [18, 19]. Поскольку митохондрии являются, образно выражаясь, энергетическими станциями клетки, при данном заболевании нарушается энергетический обмен. В условиях стресса и гипоксии энергетический обмен нарушается с преобладанием более быстрого анаэробного гликолиза, но при этом образуется только 2 молекулы АТФ, тогда как при аэробном — 38 [5]. Возникает дефицит энергии.
Такие нарушения тесно связаны с нарушениями пуринового обмена, поскольку энергия в организме хранится в виде нуклеотидов, среди которых аденин и гуанин являются пуриновыми, и они метаболизируются до мочевой кислоты, а тимин, цитозин и урацил являются пиримидиновыми и метаболизируются с образованием кетоновых тел, аммиака и β-изомасляной кислоты. Данные представления патогенетически сближают синдром циклической рвоты и синдром ацетонемической рвоты, а также объясняют необходимость и возможные пути метаболической коррекции.
Другие считают, что причиной резкого повышения кетоновых тел может быть недостаточное потребление детьми углеводов при избытке жиров и кетогенных аминокислот.
Кризы могут возникать внезапно с промежутками в несколько недель или месяцев. Провоцирующими факторами могут быть: нарушение диеты (жареные и печеные продукты), лихорадка, отказ от еды, физические и психические перегрузки.
Предвестниками синдрома циклической рвоты является анорексия, вялость или повышение возбудимости, тошнота, головные боли, абдоминальные боли, запах ацетона изо рта.
Затем появляется многократная или неукротимая рвота, которая может продолжаться от одного до пяти дней. Схваткообразные боли в животе усиливаются. Во время криза больной становится сонливым. В результате рвоты могут развиваться гемодинамические нарушения: тахикардия, мягкий пульс, приглушенность сердечных тонов, гипотония.
Печень умеренно увеличена. В некоторых случаях повышается температура. В выдыхаемом воздухе и рвотных массах ощущается запах прелых яблок. В моче высокая концентрация кетоновых тел. Приступы могут ликвидироваться спонтанно, без лечения.
Избыток кетоновых тел оказывает наркотическое действие на центральную нервную систему, что клинически проявляется вялостью, заторможенностью.
В биохимическом анализе крови обнаруживают нарушение липидного обмена (гиперхолестеринемию), тенденцию к гипогликемии, гиперкетонемию. В общем анализе крови: умеренный лейкоцитоз, нейтрофилез, ускоренная СОЭ.
В моче и выдыхаемом воздухе обнаруживается ацетон, в крови — повышенная концентрация кетоновых тел. На электроэнцефалограмме выявляются различные отклонения, не исчезающие полностью после прекращения приступа.
Этот синдром чаще встречается в дошкольном возрасте и сопровождается приступами многократной рвоты и кетонемии. У таких больных нередко выявляют повышенную возбудимость, мочекислую нефропатию, сахарный диабет, ожирение.
Кетоз при длительной рвоте, недоедании или голодании представляет классический компенсаторный процесс, призванный восполнить энергетический дефицит, точнее, недостаток углеводов, за счет альтернативных энергосубстратов кетокислот.
Диагноз синдрома ацетонемической рвоты можно подтвердить только после исключения других заболеваний, сопровождающихся рвотой: аппендицита и перитонита, энцефалитов, менингитов, начала отека головного мозга, отравления, токсикоза и инфекционных заболеваний и др. Но в первую очередь диабетического кетоацидоза.
Ацетонемические кризы у большинства детей прекращаются после 10–12 лет, но сохраняется большая вероятность развития таких патологических состояний, как подагрические кризы, вегетососудистые дистонии по гипертоническому типу, артериальная гипертензия.
Транзиторный кетоз у детей и подростков может выявляться при лихорадке, стрессах, инфекционных заболеваниях, голодании (во время болезни), употреблении богатой жирами пищи, напряженной физической активности. В этих случаях содержания кетоновых тел в моче не более 2+.
Лечение и профилактика гиперкетонемии зависят от причины ее возникновения, но во всех случаях направлены на улучшение функции печени и нормализацию энергетического обмена. Это достигается ограничением содержания жира в пищевом рационе, назначением липотропных средств (метионина и др.), витаминов группы В, при необходимости — инсулина, кокарбоксилазы.
В период приступа синдрома циклической ацетонемической рвоты выраженная дегидратация, гиповолемия, метаболический ацидоз и электролитные нарушения — это основные факторы, которые определяют тяжесть состояния. Необходимо в первую очередь ликвидировать ацидоз: назначить промывание желудка и кишечника 1–2% раствором бикарбоната натрия. Антикетогенными свойствами обладает 5–10% раствор глюкозы, с добавлением необходимого количества инсулина, а также раствор Рингера [20].
Если питье не провоцирует рвоту, рекомендуется подслащенный чай, Регидрон, Оралит — частыми и небольшими объемами. После улучшения состояния и появления возможности приема жидкости назначается кормление ребенка. Диета должна содержать легкоусвояемые углеводы и ограниченное количество жиров (манная, овсяная, гречневая каши; картофельное пюре, печеные яблоки, сухари, сухое печенье).
Итак, выяснение механизмов развития кетонемического синдрома, выделение наиболее вероятных причин формирования кетоза дают возможность установить генез заболевания, а тем самым нормализовать состояние больного и предупреждать рецидивы кетонемии.
- Березов Т. Т., Коровкин Б. Ф. Биологическая химия. Учебник. 3-е изд. М.: Медицина, 2004. 704 с.
- Stryer, Lubert. Biochemistry (Fourth ed.). New York: W. H. Freeman and Company, 1995. P. 510–515, 581–613, 775–778.
- Марри Р., Греннер Д., Мейес П., Родуэлл В. Биохимия человека. Пер. с англ. М.: Мир, 1993. Т. I. 381 с.
- Зайчик А. Ш., Чурилов Л. П. Основы патохимии. СПб: Элби-СПб, 2000. 687 с.
- Эндокринология и метаболизм. В 2 т. / Под ред. Фелинга Ф. и соавт. Пер. с англ. Кандрора В. И., Старковой Н. Т. М.: Медицина,1985. Т. 2. 416 с.
- Эндокринология: национальное руководство / Под ред. Дедова И. И., Мельниченко Г. А. М.: ГЭОТАР-Медиа, 2008.
- Лечение диабетической комы у детей. Методические рекомендации. М., 2006. 14 с.
- Brown L. M., Corrado M. M., van der Ende R. M. et al. Evaluation of glycogen storage disease as a cause of ketotic hypoglycemia in children // J Inherit Metab Dis. 2015, May; 38 (3): 489–493.
- Чибирас П. П. Гипогликемическая кетонемия как причина нейротоксикоза у детей // Вопросы охраны материнства и детства. 1982. № 2. С. 30–33.
- Генес С. Г. Гипогликемии. Гипогликемический симптомокомплекс. М.: Медицина, 1970. 236 с.
- Кроненберг Г. М. и соавт. Ожирение и нарушение липидного обмена. Пер. с англ. под ред. И. И. Дедова, Г. А. Мельниченко. М.: ООО «Рид Элсивер», 2010. 264 с.
- Лукьянчиков В. С. Кетоз и кетоацидоз. Патохимический и клинический аспект // РМЖ, 2004, т. 12, № 23, с. 1301.
- Масловская А. А. Механизм развития кетоза при сахарном диабете и голодании // Журнал Гродненского государственного медицинского университета. 2012, № 3 (39), 8–10.
- Неотложная медицинская помощь. Пер. с англ. / Под ред. Дж. Э. Тинтиналли, Р. Л. Кроума, Э. Руиза. М.: Медицина, 2001. 1000 c.
- Рябчук Ф. Н., Пирогова З. И. Коэнзимное ацетилирование и уровень свободных жирных кислот крови у детей с ацетонемией и билиарной недостаточностью // Лечащий Врач. 2012, № 8, с. 42–46.
- Li BUK: Cyclic vomiting: new understanding of an old disorder // Contemporary Ped. 1996, 13 (7): 48–62.
- Krakowczyk H., Machura E., Rusek-Zychma M., Chrobak E., Ziora K. Assessment of the natural history and clinical presentation of acetonemic vomiting. 2014, 71 (6): 323–327.
- Boles R. et al. Cyclic vomiting syndrome and mitochondrial DNA mutations // Lancet. 1997, 350: 1299–1300.
- Salpietro C. D., Briuglia S., Merlino M. V. et al. A mitochondrial DNA mutation (A3243 G mtDNA) in a family with cyclic vomiting // Am. J. Pediatr. 2003. 162. 727–728.
- Марушко Ю. В., Шев Г. Г., Полковниченко Л. Н., Мошкина Т. В. Терапевтические подходы при ацетонемическом синдроме у детей // Здоровье ребенка. 2012, № 1, с. 61–65.
В. В. Смирнов 1 , доктор медицинских наук, профессор
А. В. Симаков
ФГБОУ ВО РНИМУ им. Н. И. Пирогова МЗ РФ, Москва